基因突变分析的制作方法
基因突变分析
交叉引用
1.本技术要求于2019年7月31日提交的美国临时专利申请号62/881,180的权益,该临时申请的内容通过引用以其全文并入于此。
背景技术:
2.利用核酸扩增的研究方法,例如,下一代测序,提供了关于复杂样品、基因组和其他核酸来源的大量信息。在一些情况下,这些样品在环境中或通过基因编辑技术受到诱变条件的影响。对于涉及小样品诸如那些经受诱变条件影响的样品的研究、诊断和治疗,需要高度准确、可扩展和有效的核酸扩增和测序方法。
技术实现要素:
3.本文描述了检测样品、基因组或其他核酸来源中的突变的方法。
4.本文描述了确定突变的方法,包括:(a)将细胞群体暴露于基因编辑方法,其中,该基因编辑方法利用被配置成在靶序列中实现突变的试剂;(b)从该群体中分离出单细胞;(c)提供来自单细胞的细胞裂解物;(d)使细胞裂解物与至少一种扩增引物、至少一种核酸聚合酶和核苷酸混合物接触,其中所述核苷酸混合物包括至少一种终止子核苷酸,所述终止子核苷酸终止由所述聚合酶进行的核酸复制,(d)扩增所述靶核酸分子以生成多个终止的扩增产物,其中所述复制通过链置换复制进行;(e)将步骤(e)中获得的分子连接到衔接子上,从而生成扩增产物文库;和(f)对扩增产物文库进行测序,并将扩增产物序列与至少一种参照序列进行比较,以识别至少一个突变。本文还描述了一些方法,其中至少一个突变存在于靶序列中。本文还描述了一些方法,其中至少一个突变不存在于靶序列中。本文还描述了一些方法,其中基因编辑方法包括使用crispr、talen、zfn、重组酶、大范围核酸酶或病毒整合(有意或无意)。本文还描述了一些方法,其中基因编辑技术包括使用crispr。本文还描述了一些方法,其中基因编辑技术包括使用基因治疗方法。本文还描述了一些方法,其中基因治疗方法未配置为修饰细胞的体细胞dna或种系dna。本文还描述了一些方法,其中参照序列是基因组。本文还描述了一些方法,其中参照序列是特异性决定序列,其中该特异性决定序列被配置为与靶序列结合。本文还描述了一些方法,其中至少一个突变存在于与特异性决定序列至少有1个碱基不同的序列区域中。本文还描述了一些方法,其中至少一个突变存在于与特异性决定序列至少有2个碱基不同的序列区域中。本文还描述了一些方法,其中至少一个突变存在于与特异性决定序列至少有3个碱基不同的序列区域中。本文还描述了一些方法,其中至少一个突变存在于与特异性决定序列至少有5个碱基不同的序列区域中。本文还描述了一些方法,其中至少一个突变包含插入、缺失或置换。本文还描述了一些方法,其中参照序列是crispr rna(crrna)序列。本文还描述了一些方法,其中参照序列是单向导rna(sgrna)序列。本文还描述了一些方法,其中至少一个突变存在于与催化活性cas9结合的序列区域中。本文还描述了一些方法,其中单细胞是哺乳动物细胞。本文还描述了一些方法,其中单细胞是人类细胞。本文还描述了一些方法,其中单细胞来源于肝脏、皮
肤、肾脏、血液或肺。本文还描述了一些方法,其中单细胞是原代细胞。本文还描述了一些方法,其中单细胞是干细胞。本文还描述了一些方法,其中至少一些扩增产物包含条形码。本文还描述了一些方法,其中至少一些扩增产物包含至少两种条形码。本文还描述了一些方法,其中条形码包括细胞条形码。本文还描述了一些方法,其中条形码包括样品条形码。本文还描述了一些方法,其中至少一些扩增引物包含一种唯一分子标识符(umi)。本文还描述了一些方法,其中至少一些扩增引物包含至少两种唯一分子标识符(umi)。本文还描述了一些方法,其中该方法还包括使用pcr的额外扩增步骤。本文还描述了一些方法,其中该方法还包括在连接到衔接子之前,从终止的扩增产物中去除至少一种终止子核苷酸。本文还描述了一些方法,其中使用包括微流控装置的方法从群体中分离出单细胞。本文还描述了一些方法,其中至少一个突变发生在少于50%的细胞群体中。本文还描述了一些方法,其中至少一个突变发生在少于25%的细胞群体中。本文还描述了一些方法,其中至少一个突变发生在少于1%的细胞群体中。本文还描述了一些方法,其中至少一个突变发生在不超过0.1%的细胞群体中。本文还描述了一些方法,其中至少一个突变发生在不超过0.01%的细胞群体中。本文还描述了一些方法,其中至少一个突变发生在不超过0.001%的细胞群体中。本文还描述了一些方法,其中至少一个突变发生在不超过0.0001%的细胞群体中。本文还描述了一些方法,其中至少一个突变发生在不超过25%的扩增产物序列中。本文还描述了一些方法,其中至少一个突变发生在不超过1%的扩增产物序列中。本文还描述了一些方法,其中至少一个突变发生在不超过0.1%的扩增产物序列中。本文还描述了一些方法,其中至少一个突变发生在不超过0.01%的扩增产物序列中。本文还描述了一些方法,其中至少一个突变发生在不超过0.001%的扩增产物序列中。本文还描述了一些方法,其中至少一个突变发生在不超过0.0001%的扩增产物序列中。本文还描述了一些方法,其中至少一个突变存在于与遗传性疾病或病况相关的序列区域中。本文还描述了一些方法,其中至少一个突变存在于与dna修复酶的结合不相关的序列区域中。本文还描述了一些方法,其中至少一个突变存在于与mre11的结合不相关的序列区域中。本文还描述了一些方法,其中该方法还包括识别先前由另一种脱靶检测方法测序的假阳性突变。本文还描述了一些方法,其中该脱靶检测方法是计算机模拟预测、chip-seq、guide-seq、circle-seq、htgts(高通量全基因组易位测序)、idlv(整合缺陷型慢病毒)、digenome-seq、fish(荧光原位杂交)或discover-seq。
5.本文描述了识别特异性决定序列的方法,包括:(a)提供核酸文库,其中至少一些核酸包含特异性决定序列;(b)对至少一个细胞执行基因编辑方法,其中该基因编辑方法包括使细胞与包含至少一种特异性决定序列的试剂接触;(c)使用本文描述的方法对至少一个细胞的基因组进行测序,其中识别与至少一个细胞接触的特异性决定序列;和(d)识别提供最少脱靶突变的至少一种特异性决定序列。本文还描述了一些方法,其中该脱靶突变是同义或非同义突变。本文还描述了一些方法,其中该脱靶突变存在于基因编码区之外。
6.本文描述了体内突变分析的方法,包括:(a)对活生物体中的至少一个细胞执行基因编辑方法,其中该基因编辑方法包括使细胞与包含至少一种特异性决定序列的试剂接触;(b)从该生物体中分离出至少一个细胞;(c)使用本文描述的方法对至少一个细胞的基因组进行测序。本文还描述了一些方法,其中该方法包括至少两个细胞。本文还描述了一些方法,其还包括通过将第一细胞的基因组和第二细胞的基因组进行比较来识别突变。本文
还描述了一些方法,其中第一细胞和第二细胞来自不同的组织。
7.本文描述了预测对象年龄的方法,包括:(a)提供来自对象的至少一个样品,其中该至少一个样品包含基因组;(b)使用本文描述的方法对基因组进行测序以识别突变;(c)将步骤b中获得的突变与标准参考曲线进行比较,其中该标准参考曲线将突变计数和位置与验证的年龄相关联;和(d)基于与标准参考曲线的突变比较预测对象的年龄。本文还描述了一些方法,其中该标准参考曲线针对对象的性别是特异性的。本文还描述了一些方法,其中该标准参考曲线针对对象的种族是特异性的。本文还描述了一些方法,其中该标准参考曲线针对对象的地理位置是特异性的,该对象在该地理位置度过了其生命中的一段时期。本文还描述了一些方法,其中该对象小于50岁。本文还描述了一些方法,其中该对象小于18岁。本文还描述了一些方法,其中该对象小于15岁。本文还描述了一些方法,其中该至少一个样品大于10年。本文还描述了一些方法,其中该至少一个样品大于100年。本文还描述了一些方法,其中该至少一个样品大于1000年。本文还描述了一些方法,其中对至少2个样品进行测序。本文还描述了一些方法,其中对至少5个样品进行测序。本文还描述了一些方法,其中该至少两个样品来自不同的组织。
8.本文描述了对微生物或病毒基因组进行测序的方法,包括:(a)获得包含一种或多种基因组或基因组片段的样品;(b)使用本文描述的方法对该样品进行测序,以获得多个测序读取;和(c)对测序读取进行组装和分选,以从甚至单个细菌细胞或单个病毒颗粒生成微生物或病毒基因组。本文还描述了一些方法,其中该样品包含来自至少两种生物体的基因组。本文还描述了一些方法,其中该样品包含来自至少十种生物体的基因组。本文还描述了一些方法,其中该样品包含来自至少100种生物体的基因组。本文还描述了一些方法,其中该样品来源为包括深海喷口、海洋、矿井、溪流、湖泊、陨石、冰川或火山的环境。本文还描述了一些方法,还包括识别微生物基因组中的至少一种基因。本文还描述了一些方法,其中该微生物基因组对应于未培养的生物体。本文还描述了一些方法,其中该微生物基因组对应于共生生物体。本文还描述了一些方法,还包括在重组宿主生物体中克隆至少一种基因。本文还描述了一些方法,其中该重组宿主生物体是一种细菌。本文还描述了一些方法,其中该重组宿主生物体是埃希氏菌、芽孢杆菌或链霉菌。本文还描述了一些方法,其中该重组宿主生物体是一种真核细胞。本文还描述了一些方法,其中该重组宿主生物体是一种酵母细胞。本文还描述了一些方法,其中该重组宿主生物体是酵母或毕赤酵母。
9.本文描述了用于核酸测序的试剂盒,包括:至少一种扩增引物;至少一种核酸聚合酶;至少两种核苷酸的混合物,其中,该核苷酸的混合物包括至少一种终止子核苷酸,该终止子核苷酸终止通过聚合酶的核酸复制;和使用试剂盒进行核酸测序的说明书。本文还描述了试剂盒,其中至少一种扩增引物是随机引物。本文还描述了试剂盒,其中该核酸聚合酶是dna聚合酶。本文还描述了试剂盒,其中该dna聚合酶是链置换dna聚合酶。本文还描述了试剂盒,其中该核酸聚合酶是噬菌体phi29(φ29)聚合酶、基因修饰的phi29(φ29)dna聚合酶、dna聚合酶i的klenow片段、噬菌体m2 dna聚合酶、噬菌体phiprd1 dna聚合酶、bst dna聚合酶、bst大片段dna聚合酶、exo(-)bst聚合酶、exo(-)bca dna聚合酶、bsu dna聚合酶、vent
r dna聚合酶、ventr(exo-)dna聚合酶、deep vent dna聚合酶、deep vent(exo-)dna聚合酶、isopol dna聚合酶、dna聚合酶i、therminator dna聚合酶、t5 dna聚合酶、测序酶、t7 dna聚合酶、t7-测序酶或t4 dna聚合酶。本文还描述了试剂盒,其中该核酸聚合酶包含3
’‑
》
5’核酸外切酶活性,并且至少一种终止子核苷酸抑制该3
’‑
》5’核酸外切酶活性。本文还描述了试剂盒,其中该核酸聚合酶不包含3
’‑
》5’核酸外切酶活性。本文还描述了试剂盒,其中该聚合酶是bst dna聚合酶、exo(-)bst聚合酶、exo(-)bca dna聚合酶、bsu dna聚合酶、ventr(exo-)dna聚合酶、deep vent(exo-)dna聚合酶、klenow片段(exo-)dna聚合酶或therminator dna聚合酶。本文还描述了试剂盒,其中该至少一种终止子核苷酸包含脱氧核糖的3’碳的r基团的修饰。本文还描述了试剂盒,其中该至少一种终止子核苷酸选自于包含核苷酸的3’封闭的可逆终止子、包含核苷酸的3’未封闭的可逆终止子、包含脱氧核苷酸的2’修饰的终止子、包含对脱氧核苷酸的含氮碱基的修饰的终止子及其组合。本文还描述了试剂盒,其中该至少一种终止子核苷酸选自于双脱氧核苷酸、反向双脱氧核苷酸、3'生物素化核苷酸、3'氨基核苷酸、3'-磷酸化核苷酸、3'-o-甲基核苷酸、3'碳间隔子核苷酸(包括3'c3间隔子核苷酸)、3'c18核苷酸、3'己二醇间隔子核苷酸、无环核苷酸,及其组合。本文还描述了试剂盒,其中该至少一种终止子核苷酸选自于含有对α基团的修饰的核苷酸、c3间隔子核苷酸、锁核酸(lna)、反向核酸、2'氟核苷酸、3'磷酸化核苷酸、2'-o-甲基修饰的核苷酸和反式核酸。本文还描述了试剂盒,其中含有对α基团的修饰的核苷酸是α-硫代双脱氧核苷酸。本文还描述了试剂盒,其中该扩增引物的长度为4至70个核苷酸。本文还描述了试剂盒,其中该至少一种扩增引物的长度为4至20个核苷酸。本文还描述了试剂盒,其中该至少一种扩增引物包含随机区域。本文还描述了试剂盒,其中该随机区域的长度为4至20个核苷酸。本文还描述了试剂盒,其中该随机区域的长度为8至15个核苷酸。本文还描述了试剂盒,其中该试剂盒还包括文库制备试剂盒。本文还描述了试剂盒,其中该文库制备试剂盒包含下列中的一种或多种:至少一种多核苷酸衔接子;至少一种高保真聚合酶;至少一种连接酶;用于核酸剪切的试剂;和至少一种引物。本文还描述了试剂盒,其中该试剂盒还包含配置用于基因编辑的试剂。援引并入
10.本说明书中提到的所有出版物、专利和专利申请均通过引用并入本文,其程度如同特别地且单独地指出每一个单独的出版物、专利或专利申请均通过引用而并入。
附图说明
11.在所附权利要求书中具体阐述了本发明的新颖性特征。通过参考对在其中利用到本发明的原理的说明性实施方案加以阐述的以下详细描述和附图,将会获得对本发明的特征和优点的更好的理解,在附图中:
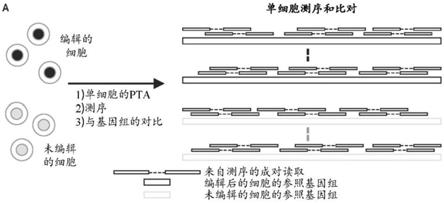
12.图1a示出了使用pta方法、单细胞测序和比对检测突变的工作流程。使用pta扩增编辑后的细胞和未编辑的对照细胞,使用短读取测序进行测序,并与参照基因组比对。
13.图1b示出了小插入/缺失的检测。通过使用变异体判定软件将比对的序列数据与参照基因组进行比较来识别插入/缺失(黑色椭圆)。通过比较编辑后的细胞和未编辑的对照细胞之间的插入/缺失,并将检索空间限制于显示与grna靶位点存在序列相似性的基因组区域,来识别可能成为crispr编辑事件的候选的插入/缺失。候选编辑事件的证据包括1)位于显示与靶位点存在相似性的基因组区域中假定的pam序列上游3-4个碱基的插入/缺失,和2)这些插入/缺失仅限于编辑后的细胞,其中在未编辑的对照细胞中没有证据。
14.图1c和1d示出了易位和大片段缺失的检测。crispr诱导的,包括染色体间和染色
体内易位、倒位和大片段缺失的结构变异体可以通过比较编辑后的和未编辑的细胞之间的双端测序映射(read-pair mapping)模式来识别。crispr诱导的易位通过编辑后的细胞中的读取对(read pair)比对来识别,其中读取对的至少两个区域与不同的染色体进行比对,并且断点位于显示与grna靶序列存在相似性的区域。这些不一致的读取对不应该存在于未编辑的细胞的比对中(图1c)。通过显示正确方向但包含与参照基因组的远侧部分对齐的区域的读取对来识别大片段缺失(图1d)。
15.图1e示出了先前的多重置换扩增(mda)方法与原代模板定向扩增(pta)方法的一个实施方案(即pta-不可逆终止子方法)的比较。
16.图1f示出了pta-不可逆终止子方法与不同实施方案(即pta-可逆终止子方法)的比较。
17.图1g示出了mda与pta-不可逆终止子方法在涉及突变传播时的比较。
18.图1h示出了扩增后进行的方法步骤,包括除去终止子、修复末端以及在衔接子连接之前进行加a尾。然后,在测序之前,可以在合并的细胞文库中对所有外显子或其他感兴趣的特定区域进行杂交介导的富集。源自每次读取的细胞均通过细胞条形码(显示为绿色和蓝色序列)识别。
19.图2a示出了在进行pta并添加浓度不断增加的终止子(上方的凝胶)后,扩增子的大小分布。下方的凝胶示出了在进行pta并添加浓度不断增加的可逆终止子后,或添加浓度不断增加的不可逆终止子后,扩增子的大小分布。
20.图2b(gc)示出了mda和pta的测序碱基的gc含量比较。
21.图2c示出了在单细胞经过pta或mda后,映射到人类基因组(p_mapped)的映射质量得分(e)(mapq)。
22.图2d在单细胞经过pta或mda后,映射到人类基因组(p_mapped)的读取百分比。
23.图2e(pcr)示出了在单细胞经过mda和pta后,2000万个亚取样读取中为pcr复制的读取百分比的比较。
24.图2f示出了扩增动力学,即mda、mda无模板对照(ntc)、pta和pta无模板对照(ntc)的扩增子产量随时间(小时)的变化。
25.图3a示出了在单细胞经过可逆或不可逆终止子pta后,映射到人类基因组(p_mapped2)的映射质量得分(c)(mapq2)。
26.图3b示出了在单细胞经过可逆或不可逆终止子pta后,映射到人类基因组(p_mapped2)的读取百分比。
27.图3c示出了使用各种方法得到的一系列箱形图,描述了与alu元件重叠的平均百分比读取的对齐读取。pta具有与基因组对齐的最大读取数。
28.图3d示出了使用各种方法得到的一系列箱形图,描述了与alu元件重叠的平均百分比读取的pcr复制。
29.图3e示出了使用各种方法得到的一系列箱形图,描述了与alu元件重叠的平均百分比读取的gc含量。
30.图3f示出了使用各种方法得到的一系列箱形图,描述了与alu元件重叠的平均百分比读取的映射质量。pta具有测试方法中最高的映射质量。
31.图3g示出了在固定的7.5x测序深度下用不同的wga方法时sc线粒体基因组覆盖宽
度的比较。
32.图4a示出了在将每个细胞下取样至4000万个配对读取后,在选择高质量mda细胞(代表~50%细胞)后,与随机引物pta扩增的细胞相比,对1号染色体上的10千碱基窗口的平均覆盖深度。该图显示mda的均匀性较差,更多窗口的覆盖深度比平均覆盖深度大(框a)或小(框c)两倍。由于高gc含量和重复区域的低映射质量,在着丝粒处mda和pta均无覆盖(框b)。
33.图4b示出了mda和pta方法的测序覆盖与基因组位置的关系图(上图)。下部箱形图示出了与大量样品(bulk sample)相比,mda和pta方法的等位基因频率。
34.图5a示出了覆盖的基因组的分数与基因组读取数目的关系图,用以评估各种方法在增加测序深度时的覆盖。pta方法在每个深度下都接近两种大量样品,这是相对于其他测试方法的改进。
35.图5b示出了基因组覆盖的变异系数与读取数目的关系图,用以评估覆盖均匀性。发现pta方法在测试方法中具有最高的均匀性。
36.图5c示出了总读取的累积分数与基因组的累积分数的洛伦兹图。发现pta方法在测试方法中具有最高的均匀性。
37.图5d示出了所测试的每种方法的计算的基尼指数的一系列箱形图,以便根据完全均匀性来评估每个扩增反应的差异。发现pta方法比其他测试方法具有可再现的更高均匀性。
38.图5e示出了判定的大量变异体的分数与读取数目的关系图。在增加测序深度时,将每种方法的变异体判定率与相应的大量样品进行比较。为了评估灵敏度,计算了在每个测序深度下,在每个细胞中发现的在被下取样至6.5亿个读取的相应大量样品中判定的变异体百分比(图5a)。pta的覆盖和均匀性的提高使得与第二最灵敏方法q-mda方法相比,检测出多30%的变异体。
39.图5f示出了与alu元件重叠的平均百分比读取的一系列箱形图。pta方法显著减少了这些杂合位点的等位基因倾斜。相对于其他测试方法,pta方法更均匀地扩增同一细胞中的两个等位基因。
40.图5g示出了变异体判定的精确度与读取数目的关系图,用以评估突变判定的精确度。使用各种方法发现的变异体若在大量样品中未发现,则被认为是假阳性。在所测试的方法中,pta方法的假阳性判定最低(精确度最高)。
41.图5h示出了对于各种方法,每种类型碱基改变的假阳性碱基改变的分数。不受理论约束,这种模式可以是依赖于聚合酶的。
42.图5i示出了对于假阳性变异体判定,与alu元件重叠的平均百分比读取的一系列箱形图。pta方法产生了假阳性变异体判定的最低等位基因频率。
43.图5j示出了使用市售试剂盒的原代白血病样品中,随着面元(bin)大小的增大的覆盖的平均变异系数(cv),以此作为对cnv判定准确性的评估。
44.图5k示出了来自单细胞的pta产物的染色体cnv图谱,其中在大量样品中判定cnv(阴影箭头)。无阴影箭头代表推断为亚克隆cnv但未在大量样品中判定的区域,其中发现五个细胞中的两个具有相同的改变。核型图中cnv检测降低的区域代表着丝粒,这表明在pta扩增的细胞中覆盖降低(对于点图和线图,误差线代表一个sd,对于箱形图,中线是中位数;
箱规格限代表上四分位数和下四分位数;箱须代表1.5倍的四分位范围;点显示离群值)。
45.图6a描绘了根据本公开内容的克隆型药物敏感性目录的示意图。通过识别不同克隆型的药物敏感性,可以创建目录,肿瘤学家可以从该目录中将在患者肿瘤中识别出的克隆型转化为最适合耐药群体的药物列表。
46.图6b示出了在100次模拟后,白血病克隆的数目随着每个克隆的白血病细胞数目增加的变化。使用每个细胞的突变率,模拟预测出随着一个细胞扩展到100至1000亿个细胞,会产生大量的小克隆(框a)。当前测序方法仅检测到频率最高的1-5个克隆(框c)。在本发明的一个实施方案中,提供了用于确定刚好低于当前方法检测水平的数百个克隆的抗药性的方法(框b)。
47.图7示出了本公开内容的示例性实施方案。与底行的诊断样品相比,未经化疗的培养选择了具有激活的kras突变的克隆(红框,右下角)。相反,该克隆被泼尼松龙或道诺霉素杀死(绿框,右上角),而频率较低的克隆则经历阳性选择(虚线框)。
48.图8是本公开内容的一个实施方案的概述,即用于量化具有特定基因型的克隆对特定药物的相对敏感性的实验设计。
49.图9(a部分)示出了具有寡核苷酸的珠子,该寡核苷酸附接有可切割的接头、唯一细胞条形码和随机引物。b部分示出了单细胞和珠子封装在同一液滴中,然后裂解细胞并切割引物。然后可以将液滴与包括pta扩增混合物的另一液滴融合。c部分示出了扩增后液滴破裂,并且合并来自所有细胞的扩增子。然后将根据本公开内容的方案用于除去终止子、末端修复和加a尾,随后连接衔接子。然后,在测序之前,使合并的细胞文库经历针对感兴趣外显子的杂交介导的富集。然后,使用细胞条形码识别源自每次读取的细胞。
50.图10a展示了使用包括细胞条形码和/或唯一分子标识符的引物将细胞条形码和/或唯一分子标识符并入pta反应中。
51.图10b展示了使用包括细胞条形码和/或唯一分子标识符的发夹引物将细胞条形码和/或唯一分子标识符并入pta反应中。
52.图11a(pta_umi)示出了唯一分子标识符(umi)的并入使得能够创建共有读取,减少了由测序和其他错误引起的假阳性率,从而导致在进行种系或体细胞变异体判定时的灵敏度提高。
53.图11b示出了将具有相同umi的读取折叠能够校正扩增和其他偏差,这些偏差在判定拷贝数变异体时可能导致错误检测或有限的灵敏度。
54.图12a示出了对于环境致突变性实验的直接测量,突变数目与治疗组的关系图。将单个人类细胞以不同的处理水平暴露于媒介物(vhc)、甘露糖(man)或直接诱变剂n-乙基-n-亚硝基脲(enu),并测量突变数目。
55.图12b示出了突变数目与不同的治疗组和水平的一系列关系图,进一步按照碱基突变的类型进行了划分。
56.图12c示出了三核苷酸背景下突变的模式表示。y轴上的碱基位于n-1位置,x轴上的碱基位于n+1位置。较暗的区域表示较低的突变频率,并且较亮的区域表示较高的突变频率。最上面一行的实心黑框(胞嘧啶突变)表明,当胞嘧啶后面是鸟嘌呤时,胞嘧啶诱变的频率降低。最下面一行的虚线黑框(胸腺嘧啶突变)表明大多数胸腺嘧啶突变发生在腺嘌呤处于胸腺嘧啶正前方时的位置。
57.图12d示出了将cd34+细胞中的已知dna酶i超敏位点的位置与n-乙基-n-亚硝基脲处理的细胞中的相应位置比较的图。没有观察到胞嘧啶变异体的显著富集。
58.图12e示出了dna酶i超敏(dh)位点中enu诱导突变的比例。以前由表观基因组学线路图计划(roadmap epigenomics project)进行分类的cd34+细胞中的dh位点被用于研究enu突变是否在代表开放染色质位点的dh位点处更普遍。在dh位点的变异体位置没有识别出显著富集,并且在dh位点没有观察到限于胞嘧啶的变异体的富集。
59.图12f示出了在具有特定注释的基因组位置中enu诱导突变的比例的一系列箱形图。相对于每个注释所包括的基因组的比例(右框),在每个细胞中的变异体(左框)的特定注释中没有看到特定的富集。
60.图13a示出了经过基因组编辑实验和pta之后,在靶位点的汉明间距7范围内,编辑后的细胞与未编辑的细胞中的插入/缺失(indel)计数。
61.图13b示出了经过基因组编辑实验和pta之后,在靶位点的汉明间距6范围内,编辑后的细胞与未编辑的细胞中的结构变异体计数。
62.图14a示出了使用pta在2个编辑后的单细胞中检测crispr诱导的编辑。
63.图14b示出了使用pta检测crispr诱导的编辑产生的大量(》1kb)缺失,该缺失仅限于编辑后的#1细胞。
64.图14c示出了使用pta在编辑后的#1细胞中检测2号染色体位置241,275,213和4号染色体位置38,536,006之间的染色体间易位。
65.图15a示出了在增加覆盖范围的测序深度时,原代白血病细胞中的比对和snv判定指标(对于每种方法,n=5,误差线代表1个sd)。
66.图15b示出了在增加cv覆盖的测序深度时,原代白血病细胞中的比对和snv判定指标(对于每种方法,n=5,误差线代表1个sd)。
67.图15c示出了在增加判定灵敏度的测序深度时,原代白血病细胞中的比对和snv判定指标(对于每种方法,n=5,误差线代表1个sd)。
68.图15d示出了在增加snv判定精确性的测序深度时,原代白血病细胞中的比对和snv判定指标(对于每种方法,n=5,误差线代表1个sd)。
69.图16a示出了同源细胞实验的概述,其中将单细胞铺板并培养,然后对个体细胞进行再分离、pta和测序。
70.图16b示出了通过将大量和单细胞数据进行比较对变异体类型进行分类的方法。
71.图16c示出了以大量细胞为标准,每个细胞的snv判定灵敏度和精确性。
72.图16d示出了对于不同的变异体类别,被判定为杂合子的变异体的百分比。
73.图16e示出了在单个cd34+人脐带血细胞中测量的假阳性和体细胞变异率。
74.图17a示出了每个样品中所有变异体的突变数概览。
75.图17b示出了每个样品中体细胞变异体的突变数概览。
76.图17c示出了每个样品中假阳性变异体的突变数概览。
77.图18a示出了种系变异体的等位基因频率分布概览。
78.图18b示出了体细胞变异体的等位基因频率分布概览。
79.图18c示出了假阳性变异体的等位基因频率分布概览。
80.图19示出了14号染色体上纯合或杂合假阳性变异体判定的密度(该染色体具有最
大数量的假阳性判定)。100kb区间的平均gc含量在该核型图下方运行。
81.图20a示出了在单细胞分辨率处测量基因组编辑策略脱靶活性的实验和计算方法,其中对单个编辑后的细胞进行测序,并且插入/缺失判定仅限于与前间隔序列有最多5个错配的位点。
82.图20b示出了每个细胞的插入/缺失判定数量。对每种对照或实验细胞类型都进行了插入/缺失判定,其中靶区域与vegfa或emx1前间隔序列存在最多5个碱基错配。图例中列出的grna或对照指定该细胞接受哪种grna。在基因组区域判定的插入/缺失与该细胞接受的grna不匹配的情况被认为是假阳性。
83.图20c示出了判定的脱靶插入/缺失位置总数的表,这些位置是一个细胞所特有的或者存在于多个细胞中。
84.图20d示出了使用emx1或vegfa grna的反复出现的插入/缺失的基因组位置。中靶位点标注为灰色。
85.图20e示出了在接受emx1或vegfa grna的每种细胞类型中识别的sv的circos圈图,其中含有跨细胞类型中出现的至少一个反复出现的断点的位点显示为绿色,或仅在该细胞类型中出现的显示为红色。每个细胞中检测到的sv数量绘制在右侧(对于箱形图,中线是中位数;箱规格限代表上四分位数和下四分位数;箱须代表1.5倍的四分位范围;点显示离群值)。
86.图21示出了去除非反复出现的单碱基对插入提高脱靶检测精确性的实验。对每种对照或实验细胞类型都进行了插入/缺失判定,要求与vegfa或emx1向导rna序列存在不超过5个错配。脱靶事件指定grna必须匹配哪个基因组区域,而在图例中列出的grna或对照指定该细胞接受哪种grna。在基因组区域判定的插入/缺失与该细胞接受的grna不匹配的情况被认为是假阳性。
87.图22a示出了使用pta方法分析的细菌样品的最长重叠群长度。
88.图22b示出了每个样品的图表,包含累计长度与累计重叠群长度的比例,以及基于与基因组序列比对的每个样品的最接近命中属。
89.图22c示出了细菌样品10的累计长度与累计重叠群长度的比例图,以及基于与嗜血杆菌和链球菌基因组序列比对的每个样品的最接近命中属。
90.图22d示出了对于每个所测试的细菌样品,与人类染色体比对的读取对。
91.图22e示出了将读取指定为人类来源的示意图。
92.图22f示出了对于所有测试的细菌样品,具有至少一个人类映射的读取的所有读取对的读取对映射位置。
93.图22g示出了属于细菌样品10的重叠群分配的分类等级。
具体实施方式
94.需要开发新的可扩展、准确且有效的核酸扩增(包括单细胞和多细胞基因组扩增)和测序方法,其将通过以可再现的方式增加序列呈现、均匀性和准确性来克服当前方法的局限性。本文提供了用于提供准确且可扩展的原代模板定向扩增(pta)和测序的组合物和方法。这些方法和组合物促进靶标(或“模板”)核酸的高精度扩增,这增加了下游应用(如下一代测序)的准确性和灵敏度。本文还提供了确定单核苷酸变异体、拷贝数变异、结构变异
体、克隆分型和环境致突变性测量的方法。通过pta测量基因组变异可用于各种应用,例如,环境致突变性、预测基因编辑技术的安全性、测量癌症治疗引起的基因组变化、测量化合物或放疗的致癌性(包括用于确定新食品或药物安全性的遗传毒性研究)、评估年龄、分析耐药细菌和在工业应用环境中识别细菌。此外,这些方法还可用于在环境条件改变后检测特定细胞群的选择,例如暴露于抗癌治疗,以及基于单个癌细胞中的突变和新抗原负荷来预测对免疫疗法的响应。
95.定义
96.除非另有定义,否则本文使用的所有技术和科学术语的含义与这些发明所属领域的普通技术人员通常所理解的含义相同。
97.在整个本公开内容中,数字特征以范围格式表示。应理解,范围格式的描述仅为了方便和简洁,而不应被解释为对任何实施方案的范围的严格限制。因此,除非上下文另有明确指示,否则应认为对范围的描述已具体公开了所有可能的子范围以及在该范围内直至下限单位的十分之一的单个数值。例如,对范围如从1至6的描述应视为已具体公开了子范围,如从1至3、从1至4、从1至5、从2至4、从2至6、从3至6等,以及该范围内的单个值,例如,1.1、2、2.3、5和5.9。无论范围的宽度如何,这都适用。这些中间范围的上限和下限可以独立地包括在较小的范围内,并且也包括在本发明内,受规定范围内任何明确排除的限制。当所述范围包括一个或两个限值时,除非上下文另外明确指出,否则不包含所包括的那些限值中的一个或两个的范围也包括在本发明中。
98.本文所使用的术语仅出于描述特定实施方案的目的,并且不旨在限制任何实施方案。如本文所用,单数形式“一”、“一个”和“该”也旨在包括复数形式,除非上下文另有明确指出。还将理解,当在本说明书中使用术语“包括(comprises)”和/或包含(comprising)时,其指定了所述特征、整体、步骤、操作、元素和/或组件的存在,但并不排除一个或多个其他特征、整体、步骤、操作、元素、组件和/或其群组的存在或添加。如本文所用,术语“和/或”包括一个或多个相关联的所列项目的任何和所有组合。
99.除非特别说明或从上下文可以明显看出,否则如本文所用,术语“约”在提及数值或数值范围时应理解为是指所述数值+/-其10%,或对于一个范围中列出的值,则指从比列出的下限低10%到比列出的上限高10%。
100.如本文所用,术语“对象”或“患者”或“个体”是指动物,包括哺乳动物,如人类、兽医动物(例如,猫、狗、牛、马、绵羊、猪等)和疾病实验动物模型(例如,小鼠、大鼠)。根据本发明,可以在本领域技术范围内使用常规分子生物学、微生物学和重组dna技术。这些技术在文献中有充分的解释。参见,例如,sambrook,fritsch和maniatis,molecular cloning:a laboratory manual,第二版(1989)cold spring harbor laboratory press,cold spring harbor,new york(本文中为"sambrook等人,1989");dna cloning:a practical approach,第i和ii卷(d.n.glover著,1985);oligonucleotide synthesis(mj.gait著,1984);nucleic acid hybridization(b.d.hames和s.j.higgins著,(1985));transcription and translation(b.d.hames和s.j.higgins著,(1984));animal cell culture(r.i.freshney著,(1986));immobilized cells and enzymes(lrl出版社(1986));b.perbal,a practical guide to molecular cloning(1984);f.m.ausubel等人(著),current protocols in molecular biology,john wiley&sons,inc.(1994);等等。
101.术语“核酸”涵盖多链以及单链分子。在双链或三链核酸中,核酸链不需要是共延伸的(即,双链核酸不需要沿两条链的整个长度是双链的)。本文所述的核酸模板可以根据样品(从小的无细胞dna片段至整个基因组)而具有任何大小,包括但不限于长度为50-300个碱基、100-2000个碱基、100-750个碱基、170-500个碱基、100-5000个碱基、50-10,000个碱基或50-2000个碱基。在一些情况下,模板的长度为至少50、100、200、500、1000、2000、5000、10,000、20,000、50,000、100,000、200,000、500,000、1,000,000或大于1,000,000个碱基。本文所述的方法提供了核酸如核酸模板的扩增。本文所述的方法另外提供了分离的和至少部分纯化的核酸以及核酸文库的生成。核酸包括但不限于dna、rna、环状rna、cfdna(无细胞dna)、cfrna(无细胞rna)、sirna(小干扰rna)、cffdna(无细胞胎儿dna)、mrna、trna、rrna、mirna(微rna)、合成多核苷酸、多核苷酸类似物,符合本说明书的任何其他核酸,或其任何组合。当提供多核苷酸时,其长度用碱基数和缩写描述,如nt(核苷酸)、bp(碱基)、kb(千碱基)或gb(千兆碱基)。
102.如本文所用,术语“液滴”是指液滴致动器上的一定体积液体。在一些情况下,例如,液滴是水性或非水性的,或者可以是包括水性和非水性组分的混合物或乳液。对于可经受液滴操作的液滴流体的非限制性示例,参见,例如,国际专利申请公开号wo2007/120241。在本文提出的实施方案中,可以使用任何适于形成和操纵液滴的系统。例如,在一些情况下,使用液滴致动器。对于可以使用的液滴致动器的非限制性示例,参见,例如,美国专利号6,911,132、6,977,033、6,773,566、6,565,727、7,163,612、7,052,244、7,328,979、7,547,380、7,641,779、美国专利申请公开号us20060194331、us20030205632、us20060164490、us20070023292、us20060039823、us20080124252、us20090283407、us20090192044、us20050179746、us20090321262、us20100096266、us20110048951、国际专利申请公开号wo2007/120241。在一些情况下,珠子在液滴中、在液滴操作间隙中或在液滴操作表面上提供。在一些情况下,珠子在位于液滴操作间隙外部或与液滴操作表面分开的储器中提供,并且该储器可以与流动路径相关联,该流动路径允许包括该珠子的液滴进入液滴操作间隙或与液滴操作表面接触。用于固定磁响应珠子和/或非磁响应珠子和/或使用珠子进行液滴操作方案的液滴致动器技术的非限制性示例在美国专利申请公开号us20080053205、国际专利申请公开号wo2008/098236、wo2008/134153、wo2008/116221、wo2007/120241中描述。珠子特性可以在本文所述的方法的多路复用实施方案中采用。具有适合于多路复用的特性的珠子的示例,以及检测和分析从这种珠子发出的信号的方法,可以在美国专利申请公开号us20080305481、us20080151240、us20070207513、us20070064990、us20060159962、us20050277197、us20050118574中找到。
103.如本文所用,术语“唯一分子标识符(umi)”是指附接于多个核酸分子中的每一个的唯一核酸序列。当并入核酸分子中时,在一些情况下,umi被用于通过直接对扩增后测序的umi进行计数来校正后续的扩增偏倚。umi的设计、并入和应用在例如国际专利申请公开号wo 2012/142213、islam等人.nat.methods(2014)11:163-166;kivioja,t.等人,nat.methods(2012)9:72-74;brenner等人(2000)pnas 97(4),1665以及hollas和schuler,(2003)会议:第三届生物信息学算法国际研讨会,第2812卷中描述。
104.如本文所用,术语“条形码”是指可用于识别核酸材料的样品或来源的核酸标签。因此,在一些情况下,在核酸样品来自多个来源的情况下,每个核酸样品中的核酸用不同的
核酸标签标记,从而可以识别样品的来源。条形码,通常也称为索引、标签等,是本领域技术人员熟知的。任何合适的条形码或条形码组都可以使用。参见,例如,美国专利号8,053,192和国际专利申请公开号wo2005/068656中提供的非限制性示例。单细胞的条形码化可以例如如美国专利申请公开号2013/0274117中描述地进行。
105.本文中的术语“固体表面”、“固体支持物”和其他语法等同物是指适合于或可以被修饰以适合于本文所述的引物、条形码和序列的附接的任何材料。示例性基底包括但不限于玻璃和改性或功能化玻璃、塑料(包括丙烯酸、聚苯乙烯,和苯乙烯与其他材料的共聚物、聚丙烯、聚乙烯、聚丁烯、聚氨酯、等)、多糖、尼龙、硝酸纤维素、陶瓷、树脂、二氧化硅、二氧化硅基材料(例如,硅或改性硅)、碳、金属、无机玻璃、塑料、光纤束和各种其他聚合物。在一些实施方案中,固体支持物包括适于以有序模式固定引物、条形码和序列的图案化表面。
106.如本文所用,术语“生物样品”包括但不限于组织、细胞、生物流体及其分离物。在一些情况下,本文所述方法中使用的细胞或其他样品是从人类患者、动物、植物、土壤或包括如细菌、真菌、原生动物等微生物的其他样品中分离的。在一些情况下,生物样品来源于人类。在一些情况下,生物样品并非来源于人类。在一些情况下,细胞经历本文所述的pta方法和测序。在整个基因组或特定位置检测到的变异体可以与从该对象分离的所有其他细胞进行比较,以追踪细胞谱系的历史,以用于研究或诊断目的。
107.在一些情况下,术语“精确性”和“特异性”被当作同义词使用。在一些情况下,精确性(或阳性预测值)定义了真实的阳性命中数量除以识别的阳性命中总数(真阳性数+假阳性数)。
108.当提及聚合酶介导的扩增反应时,术语“循环”在本文中用于描述双链核酸的至少一部分的解离步骤(例如,来自扩增子的模板或双链模板的变性)、至少一部分引物与模板的杂交(退火)和引物的延伸以生成扩增子。在一些情况下,温度在扩增循环(例如等温反应)中保持恒定。在一些情况下,循环次数与生成的扩增子数量正相关。在一些情况下,等温反应的循环次数是由允许反应进行的时间量来控制的。
109.方法和应用
110.本文描述了使用pta方法识别细胞突变的方法。在一些情况下使用pta方法会导致对已知方法(例如,mda)的改进。在一些情况下,与mda方法相比,pta的假阳性和假阴性变异体判定率较低。在一些情况下,将基因组,如na12878铂基因组,用于确定pta的更大的基因组覆盖和均匀性是否会导致较低的假阴性变异体判定率。不受理论的约束,可以确定pta中错误传播的缺失降低了假阳性变异体判定率。在一些情况下,通过比较已知阳性基因座处杂合突变判定的等位基因频率来评估用这两种方法在等位基因之间的扩增平衡。在一些情况下,通过pcr进一步扩增使用pta生成的扩增子文库。在一些情况下,pta方法识别群体的单细胞中存在的突变,其中,由pta检测到的突变发生在群体中少于2%、1%、0.5%、0.2%、0.1%、0.05%、0.02%、0.01%、0.001%、0.0001%或少于0.00001%的细胞中。在一些情况下,对于给定的碱基或区域,pta方法识别小于2%、1%、0.5%、0.2%、0.1%、0.05%、0.02%、0.01%、0.001%、0.0001%或小于0.00001%的测序读数中的突变。
111.基因编辑的安全性
112.从修正导致或促成疾病(例如,镰状细胞性贫血和许多其他疾病)形成的基因到根
除目前无法治愈的感染性疾病,基因组编辑工具的持续发展显示出对改善人类健康的巨大前景。然而,这些干预的安全性仍然不清楚,因为我们还不完全了解这些工具如何与编辑细胞基因组中的其他位置相互作用并永久改变其他位置。已经开发了评估基因组编辑策略的脱靶率的方法,但迄今为止已开发的工具都是一起质询(interrogate)细胞群,导致无法测量每个细胞的脱靶率和细胞间脱靶活性的差异,也无法检测发生在少数细胞中的罕见编辑事件。这些测量基因组编辑保真度的次优策略导致用于确定给定基因组编辑方法的灵敏度和精确性的有限能力。
113.基因治疗方法可包括修饰突变的致病基因、敲除致病基因或在细胞中引入新基因。在一些情况下,这些方法包括基因组dna的修饰。在其他情况下,将病毒或其他递送系统配置为使得它们不会整合或修饰细胞中的基因组dna。然而,这样的系统仍然可能对体细胞或种系dna生成不需要的或非预期的修饰。利用pta提高单细胞中的变异体判定灵敏度和精确性,在一些情况下,对单细胞中具有高灵敏度的基因治疗方法的非预期插入率进行定量测量。在一些情况下,该方法可以通过检测周围序列来检测特定序列在非期望位置的插入,以确定基因治疗方法是否导致宿主基因组的插入或修饰。
114.本文描述了识别经历过基因组编辑(例如,crispr(簇状规律间隔性短回文重复序列)、talen(转录激活因子样效应物核酸酶)、zfn(锌指核酸酶)、重组酶、大范围核酸酶、病毒整合或其他基因组编辑技术)的动物、植物或微生物细胞中的突变和结构修饰(即易位、插入和缺失)的方法。在一些实施方案中,基因组编辑是无意的,或者是另一个过程的次级效应。在一些情况下,基因组编辑包括位点特异性或靶向基因组编辑。在一些情况下,可以分离出此类细胞,并进行pta和测序,以确定每个细胞中的突变负荷、突变组合和结构变异体。在一些情况下,由基因组编辑方案产生的每细胞突变率和突变位置用于评估给定基因组编辑方法的安全性和/或效率。在一些情况下,突变的识别包括将使用pta方法获得的测序数据与参照序列进行比较。在一些情况下,该参照序列是基因组。在一些情况下,在基因编辑过程后,由pta识别出至少一个突变。在一些情况下,参照序列是特异性决定序列,其促进向核酸的靶序列中引入突变。在一些情况下,在基因编辑过程后,由pta识别出至少一个突变,其中该突变位于靶序列中。在一些情况下,通过识别至少一个不在靶序列中的突变来分析脱靶突变率。尽管基于与靶序列的序列同源性,可以预测核酸的一些区域会发生脱靶突变,但是具有较低同源性的区域也可能具有脱靶突变。在一些情况下,pta方法识别序列的脱靶区域中的突变,该序列包含与靶序列或其反向互补序列的至少3、4、5、6、7或8个碱基错配。在一些情况下,用pta分析单细胞。在一些情况下,用pta分析细胞群体。
115.目前的许多突变分析方法都获得对批量细胞群体的测序数据。然而,这些方法提供了有限的关于群体中实际突变频率的信息,在一些情况下使用pta的单细胞分析提供了对插入脱靶率、链断裂(导致突变)和易位的更高的分辨率,因为细胞(即单细胞)的数量是已知的。pta在已知数量的单细胞中具有已知的变异检测率,在一些情况下,这允许该方法准确地识别细胞群体中每个细胞的频率和改变的组合。在一些情况下,使用pta分析至少10、100、1000、10,000、100,000个或超过100,000个单细胞,以确定变异率。在一些情况下,使用pta分析不超过10、100、1000、10,000、100,000个或不超过100,000个单细胞,以确定变异率。在一些情况下,使用pta分析10-1000、50-5000、100-100,000、1000-100,000、100-1,000,000或100-10,000个单细胞,以确定变异率。在一些情况下,从细胞群体的批量测序中
不会识别或检测出通过分析一个或多个单细胞识别的突变。
116.crispr可用于将突变引入一种或多种细胞,如哺乳动物细胞,然后通过pta来分析突变。在一些情况下,特异性决定序列存在于crispr rna(crrna)或单向导rna(sgrna)中。在一些情况下,哺乳动物细胞是人类细胞。在一些情况下,细胞来源于肝脏、皮肤、肾脏、血液或肺。在一些情况下,细胞是原代细胞。在一些情况下,细胞是干细胞。以前报道的识别从crispr生成的脱靶突变的方法包括下拉与催化活性cas9结合的序列,然而这可能导致假阳性,因为并非所有cas9结合位点都会引入突变。在一些情况下,pta方法识别存在于与催化活性cas9结合的序列区域中的至少一个突变。在一些情况下,对存在于与催化活性cas9结合的序列区域中的至少一个突变,pta方法产生更少的假阳性。
117.本文描述了识别经历过基因组编辑(例如,crispr、talen、zfn、重组酶、大范围核酸酶、病毒整合或其他技术)的动物、植物或微生物细胞中的突变的方法,其中,该方法包括在至少一种终止子核苷酸存在下扩增基因组或其片段。在一些情况下,具有终止子的扩增是在溶液中进行的。在一些情况下,将至少一个引物或至少一个基因组片段中的一个附着到表面上。在一些情况下,将至少一个引物附着于第一固体支持物,并将至少一个基因组片段附着于第二固体支持物,其中,第一固体支持物和第二固体支持物未连接。在一些情况下,将至少一个引物附着于第一固体支持物,并将至少一个基因组片段附着于第二固体支持物,其中,第一固体支持物和第二固体支持物不是同一固体支持物。在一些情况下,该方法包括在至少一种终止子核苷酸存在下扩增基因组或其片段,其中,扩增循环次数小于12、10、9、8、7、6、5、4或小于3个循环。在一些情况下,扩增产物的平均长度为100-1000、200-500、200-700、300-700、400-1000或500-1200个碱基。在一些情况下,该方法包括在至少一种终止子核苷酸存在下扩增基因组或其片段,其中,扩增循环次数不超过6个循环。在一些情况下,该至少一种终止子核苷酸确实包含可检测标记或标签。在一些情况下,该扩增包含2、3或4个终止子核苷酸。在一些情况下,至少两个终止子核苷酸包含不同的碱基。在一些情况下,至少三个终止子核苷酸包含不同的碱基。在一些情况下,四个终止子核苷酸各自包含不同的碱基。
118.本文描述了用于确定基因疗法安全性的方法。在一些情况下,通过基因编辑或其他表达方法改变细胞的功能。在一些情况下,对改变细胞功能的病毒递送系统进行配置,使其不会整合到细胞的基因组中。在一些情况下,pta方法用于识别细胞基因组的非预期的或不需要的变化。在一些情况下,pta用于识别由基因疗法造成的对体细胞或种系dna的突变。
119.肿瘤细胞的克隆分析
120.在一些情况下,使用本文所述的方法分析的细胞包括肿瘤细胞。例如,循环肿瘤细胞可以从取自患者的体液如但不限于血液、骨髓、尿液、唾液、脑脊髓液、胸膜液、心包液、腹水或房水中分离。然后,使细胞经历本文所述的方法(例如,pta)并测序,以确定每个细胞的突变负荷和突变组合。在一些情况下,这些数据用于诊断特定疾病或用作预测治疗应答的工具。类似地,在一些情况下,恶性潜能未知的细胞是从取自患者的体液如但不限于血液、骨髓、尿液、唾液、脑脊髓液、胸膜液、心包液、腹水、房水、囊胚腔液或培养物中的细胞周围的收集介质中分离的。在一些情况下,样品是从胚胎细胞周围的收集介质中获得的。在利用本文所述的方法和测序后,这些方法还用于确定每个细胞的突变负荷和突变组合。在一些情况下,这些数据用于诊断特定疾病或用作预测恶性前状态发展为显性恶性肿瘤的工具。
在一些情况下,可以从原发性肿瘤样品中分离细胞。然后,可以对细胞进行pta和测序,以确定每个细胞的突变负荷和突变组合。这些数据可以用于诊断特定疾病或用作预测患者恶性肿瘤对可用抗癌药物的抗药性的工具。通过将样品暴露于不同的化疗药物,已发现主要和次要克隆对特定药物具有不同的敏感性,这些敏感性不一定与已知的“驱动突变”的存在相关,这表明克隆种群中的突变组合决定了它对特定化疗药物的敏感性。不受理论的约束,这些发现表明,如果检测到尚未扩展并且可演化为基因组修饰数目增加的克隆而使其更有可能对治疗产生抗性的癌前病变,则可能更容易根除该恶性肿瘤。参见,ma等人,2018,“pan-cancer genome and transcriptome analyses of 1,699pediatric leukemias and solid tumors”。在一些情况下,单细胞基因组学方案用于检测从患者样品中分离的正常和恶性细胞混合物内的单个癌细胞或克隆型中的体细胞遗传变异体组合。在一些情况下,该技术还用于识别在体外和/或患者体内暴露于药物后经历阳性选择的克隆型。如图6a所示,通过比较暴露于化疗的存活克隆与诊断时识别的克隆,可以创建癌症克隆型目录,该目录记录它们对特定药物的抗性。在一些情况下,pta方法检测由多种克隆型组成的样品中的特定克隆对现有药物或新药及其组合的敏感性,其中该方法可以检测特定克隆对药物的敏感性。在一些情况下,这种方法显示了药物对特定克隆的功效,而当前的药物敏感性测量在一次测量中考虑了所有癌症克隆的敏感性,因此可能无法检测到这种功效。当将本文所述的pta应用于诊断时收集的患者样品以检测给定患者癌症中的癌症克隆型时,可以随后使用药物敏感性目录来查找这些克隆,从而告知肿瘤学家哪种药物或药物组合无效,哪种药物或药物组合最有可能对患者的癌症有效。pta可用于分析包含细胞群的样品。在一些情况下,样品包含神经元或神经胶质细胞。在一些情况下,样品包括细胞核。
121.临床和环境诱变
122.本文描述了测量环境因子致突变性的方法。例如,细胞(单细胞或细胞群体)暴露于潜在的环境条件。例如,在一些情况下,该方法使用诸如源自器官(肝脏、胰腺、肺、结肠、甲状腺或其他器官)、组织(皮肤或其他组织)、血液或其他生物来源的细胞。在一些情况下,环境条件包括热、光(例如,紫外线)、辐射、化学物质或其任何组合。在一些情况下,在一定量的环境条件暴露后,这是几分钟、几小时、几天或更长时间,分离出单细胞并进行pta方法。在一些情况下,分子条形码和唯一分子标识符用于标记样品。对样品进行测序,然后进行分析,以识别暴露于环境条件所导致的突变。在一些情况下,将这种突变与对照环境条件如已知的非诱变物质、媒介物/溶剂或缺乏环境条件进行比较。在一些情况下,这种分析不仅提供了由环境条件引起的突变总数,而且还提供了这种突变的位置和性质。在一些情况下,模式从数据中识别,并且可以用于诊断疾病或病况。在一些情况下,模式可用于预测未来的疾病状态或病况。在一些情况下,本文所述的方法测量在暴露于环境药剂,例如,潜在的诱变剂或致畸剂后细胞的突变负荷、位置和模式。在一些情况下,该方法用于评估给定药剂的安全性,包括其诱发可能导致疾病发展的突变的可能性。例如,该方法可用于预测暴露于特定浓度的特定药剂后该药剂对特定细胞类型的致癌性或致畸性。在一些情况下,该药剂是药品或药物。在一些情况下,该药剂是食物。在一些情况下,该药剂是基因改造食物。在一些情况下,该药剂是杀虫剂或其他农业化学品。在一些情况下,使用突变的位置和频率来预测生物体的年龄。在一些情况下,这种方法是在数百年、数千年或数万年前的样品上进行的。在一些情况下,将突变模式与其他数据方法(如碳年代测定法)进行比较,以生成标准曲
线。在一些情况下,人类的年龄是通过比较样品中的突变数量和模式来确定的。
123.本文描述了确定用于细胞治疗的细胞中的突变的方法,该细胞治疗诸如但不限于诱导多能干细胞的移植,尚未被操纵的造血细胞或其他细胞的移植,或经过基因组编辑的造血细胞或其他细胞的移植。然后,细胞可以经历pta和测序,以确定每个细胞的突变负荷和突变组合。细胞治疗产品中的每细胞突变率和突变位置可用于评估包括新抗原负荷的测量的产品的安全性和潜在功效。
124.微生物样品
125.本文描述了分析微生物样品的方法。在另一个实施方案中,微生物细胞(例如,细菌、真菌、原生动物)可以从植物或动物(例如,微生物群样品[例如,gi微生物群、皮肤微生物群等]或体液,例如,血液、骨髓、尿液、唾液、脑脊髓液、胸膜液、心包液、腹水或房水)分离。此外,微生物细胞可以从留置的医疗装置,如但不限于,静脉导管、导尿管、脑脊髓分流器、假体瓣膜、人工关节或气管导管分离。然后,细胞可以经历pta和测序,以确定特定微生物的身份,并检测预测对特定抗菌剂的应答(或抗性)的微生物遗传变异体的存在。这些数据可用于诊断特定的传染病和/或用作预测治疗应答的工具。在一些情况下,分析单个微生物细胞的突变。在一个实施方案中,pta用于识别具有高工业应用价值例如生产生物燃料或环境修复(溢油清理、co2封存/清除)的微生物。在一些情况下,从极端环境例如深海喷口、海洋、矿井、溪流、湖泊、陨石、冰川或火山中获取微生物样品。在一些情况下,微生物样品包括在实验室标准条件下“未培养”的微生物菌株。在一些情况下,使用pta制备的微生物样品的测序包括获得用于组装成重叠群的测序读取。在一些情况下,获得不超过10万、50万、100万、150万、200万、300万、500万、800万或1000万个读取。在一些情况下,微生物样品的分析和识别包括将组装的重叠群与已知的微生物基因组参照序列进行比较。在一些情况下,最大的组装重叠群用于与参照序列进行比较。在一些情况下,对映射到人类基因组dna中的一个或多个基因的读取进行过滤。在一些情况下,如果两个读取(前向和后向)都映射到人类基因,则进行过滤。在一些情况下,如果至少一个读取(前向和后向)映射到人类基因,则进行过滤。在一些情况下,人类基因是grch38。在一些情况下,将免组装识别方法与pta一起使用。在一些情况下,使用了免组装方法,例如kraken。在一些情况下,免组装方法包括使用参考数据库根据k-mers将读取分配给分类群。
[0126]
胎儿细胞
[0127]
用于pta方法的细胞可以是胎儿细胞,例如胚胎细胞。在一些实施方案中,将pta与非侵入性植入前遗传测试(nipgt)一起使用。在又一实施方案中,可以从体外受精产生的卵裂球或胚细胞中分离出细胞。然后,可以对细胞进行pta(例如,用pta扩增细胞中的核酸)和测序,以确定每个细胞中潜在疾病易感性遗传变异体的负荷和组合。然后可以将细胞的突变谱用于在植入前推断卵裂球对特定疾病的遗传易感性。在一些情况下,培养中的胚胎脱落核酸,这些核酸用于通过低通基因组测序来评估该胚胎的健康状况。在一些情况下,对胚胎进行冷冻-解冻。在一些情况下,从胚细胞培养条件培养基(bccm)、囊胚腔液(bf)或其组合中获得核酸。在一些情况下,胎儿细胞的pta分析用于检测染色体异常,如胎儿非整倍性。在一些情况下,pta用于检测疾病,例如唐氏综合征或帕套综合征。在一些情况下,将冷冻的胚细胞解冻并培养一段时间,然后获取核酸进行分析(例如,培养基、bf或细胞活检)。在一些情况下,培养胚细胞不超过4、6、8、12、16、24、36、48小时或不超过64小时,之后获取核酸
进行分析。
[0128]
突变
[0129]
在一些情况下,本文描述的方法(例如pta)产生了对突变检测的更高检测灵敏度和/或更低假阳性率。在一些情况下,突变是所分析序列(例如,使用本文描述的方法)和参照序列之间的差异。在一些情况下,参照序列获自其他生物体、同一或类似物种的其他个体、生物体群体或同一基因组的其他区域。在一些情况下,在质粒或染色体上识别突变。在一些情况下,突变是snv(单核苷酸变异)、snp(单核苷酸多态性)或cnv(拷贝数变异或cna/拷贝数畸变)。在一些情况下,突变是碱基置换、插入或缺失。在一些情况下,突变是转换、颠换、无义突变、沉默突变、同义突变或非同义突变、非致病突变、错义突变或移码突变(缺失或插入)。在一些情况下,当与一些方法,例如计算机模拟预测、chip-seq、guide-seq、circle-seq、htgts(高通量全基因组易位测序)、idlv(整合缺陷型慢病毒)、digenome-seq、fish(荧光原位杂交)或discover-seq相比时,pta产生了对于突变检测更高的检测灵敏度和/或更低的假阳性率。
[0130]
原代模板定向扩增
[0131]
本文描述了核酸扩增方法,如“原代模板定向扩增(pta)”。例如,图1a-图1h示意性地呈现了本文所述的pta方法。在pta方法中,使用聚合酶(例如,链置换聚合酶)优先从原代模板(“直接拷贝”)生成扩增子。因此,与mda相比,在随后的扩增过程中,错误以较低的速率从子扩增子传播。因而得到一种易于执行的方法,与现有的wga方案不同,该方法可以以准确且可再现的方式扩增低输入量的dna(包括单细胞的基因组),且具有高覆盖范围和均匀性。此外,终止的扩增产物可以在除去终止子后进行定向连接,允许细胞条形码附接至扩增引物,从而可以在进行平行扩增反应后合并来自所有细胞的产物(图1f)。在一些情况下,在扩增和/或衔接子连接之前,不需要去除终止子。
[0132]
本文描述了使用具有链置换活性的核酸聚合酶进行扩增的方法。在一些情况下,这种聚合酶具有链置换活性和低错误率。在一些情况下,这种聚合酶具有链置换活性和校正核酸外切酶活性,如3
’‑
》5’校正活性。在一些情况下,核酸聚合酶与其他组分,如可逆或不可逆终止子,或其他链置换因子结合使用。在一些情况下,聚合酶具有链置换活性,但不具有核酸外切酶校正活性。例如,在一些情况下,这些聚合酶包括噬菌体phi29(φ29)聚合酶,其也有非常低的错误率,这是3
’‑
》5’校正核酸外切酶活性的结果(参见,例如,美国专利号5,198,543和5,001,050)。在一些情况下,链置换核酸聚合酶的非限制性示例包括,例如,基因修饰的phi29(φ29)dna聚合酶、dna聚合酶i的klenow片段(jacobsen等人,eur.j.biochem.45:623-627(1974))、噬菌体m2 dna聚合酶(matsumoto等人,gene 84:247(1989))、噬菌体phiprd1 dna聚合酶(jung等人,proc.natl.acad.sci.usa84:8287(1987);zhu和ito,biochim.biophys.acta.1219:267-276(1994))、bst dna聚合酶(例如,bst大片段dna聚合酶(exo(-)bst;aliotta等人,genet.anal.(netherlands)12:185-195(1996))、exo(-)bca dna聚合酶(walker和linn,clinical chemistry 42:1604-1608(1996))、bsu dna聚合酶、包括ventr(exo-)dna聚合酶的vent
r dna聚合酶(kong等人,j.biol.chem.268:1965-1975(1993))、包括deep vent(exo-)dna聚合酶的deep vent dna聚合酶、isopol dna聚合酶、dna聚合酶i、therminator dna聚合酶、t5 dna聚合酶(chatterjee等人,gene 97:13-19(1991))、测序酶(us.biochemicals)、t7 dna聚合酶、t7-测序酶、t7 gp5 dna聚合酶、
prdi dna聚合酶、t4 dna聚合酶(kaboord和benkovic,curr.biol.5:149-157(1995))。另外的链置换核酸聚合酶也与本文所述的方法相容。给定聚合酶进行链置换复制的能力可以被确定,例如,通过在链置换复制测定中使用该聚合酶(例如,如美国专利号6,977,148中所公开)。在一些情况下,这些测定是在适合于所用酶的最佳活性温度下进行的,例如,phi29 dna聚合酶的该温度为32℃,exo(-)bst dna聚合酶的该温度为46℃至64℃,或来自超高温生物的酶的该温度为约60℃至70℃。选择聚合酶的另一种有用的测定法是在kong等人,j.biol.chem.268:1965-1975(1993)中所述的引物阻断测定。该测定包括在存在或不存在寡核苷酸的情况下使用m13 ssdna模板进行的引物延伸测定,该寡核苷酸在延伸引物的上游杂交,以阻断其进程。在该测定中,在一些情况下能够置换阻断引物的其他酶对所公开的方法有用。在一些情况下,聚合酶以近似相等的比率并入dntp和终止子。在一些情况下,本文所述聚合酶的dntp和终止子的并入比率为约1:1、约1.5:1、约2:1、约3:1、约4:1、约5:1、约10:1、约20:1、约50:1、约100:1、约200:1、约500:1或约1000:1。在一些情况下,本文所述聚合酶的dntp和终止子的并入比率为1:1至1000:1、2:1至500:1、5:1至100:1、10:1至1000:1、100:1至1000:1、500:1至2000:1、50:1至1500:1或25:1至1000:1。
[0133]
本文描述了扩增方法,其中可以通过使用链置换因子例如解旋酶来促进链置换。在一些情况下,这些因子与另外的扩增组分,如聚合酶、终止子或其他组分结合使用。在一些情况下,链置换因子与不具有链置换活性的聚合酶一起使用。在一些情况下,链置换因子与具有链置换活性的聚合酶一起使用。不受理论的约束,链置换因子可以增加较小的双链扩增子被引发的速率。在一些情况下,可以在存在链置换因子的情况下进行链置换复制的任何dna聚合酶都适用于pta方法,即使该dna聚合酶在不存在这种因子的情况下无法进行链置换复制。在一些情况下,可用于链置换复制的链置换因子包括(但不限于)bmrf1聚合酶辅助亚基(tsurumi等人,j.virology 67(12):7648-7653(1993));腺病毒dna结合蛋白(zijderveld和van der vliet,j.virology 68(2):1158-1164(1994));单纯疱疹病毒蛋白icp8(boehmer和lehman,j.virology 67(2):711-715(1993);skaliter和lehman,proc.natl.acad.sci.usa 91(22):10665-10669(1994));单链dna结合蛋白(ssb;rigler和romano,j.biol.chem.270:8910-8919(1995));噬菌体t4基因32蛋白(villemain和giedroc,biochemistry 35:14395-14404(1996);t7解旋酶-引发酶;t7gp2.5 ssb蛋白;tte-uvrd(来自腾冲嗜热厌氧菌(thermoanaerobacter tengcongensis));小牛胸腺解旋酶(siegel等人,j.biol.chem.267:13629-13635(1992));细菌ssb(例如,大肠杆菌ssb);真核生物中的复制蛋白a(rpa);人类线粒体ssb(mtssb)和重组酶(例如,重组酶a(reca)家族蛋白、t4 uvsx、噬菌体hk620的sak4、rad51、dmc1或radb)。促进链置换和引发的因子组合也符合本文所述方法。例如,解旋酶与与聚合酶结合使用。在一些情况下,pta方法包括使用单链dna结合蛋白(ssb、t4 gp32或其他单链dna结合蛋白)、解旋酶和聚合酶(例如,saudna聚合酶、bsu聚合酶、bst2.0、gspm、gspm2.0、gspssd或其他合适的聚合酶)。在一些情况下,逆转录酶与本文所述的链置换因子结合使用。在一些情况下,使用聚合酶和切口酶(如“near”)例如us 9,617,586中描述的那些酶进行扩增。在一些情况下,该切口酶是nt.bspqi、nb.bbvci、nb.bsmi、nb.bsrdi、nb.btsi、nt.alwi、nt.bbvci、nt.bstnbi、nt.cvipii、nb.bpu10i或nt.bpu10i。
[0134]
本文描述了扩增方法,其包括使用终止子核苷酸、聚合酶和其他因子或条件。例
如,在一些情况下,这些因子在扩增过程中用于使核酸模板或扩增子片段化。在一些情况下,这些因子包括核酸内切酶。在一些情况下,因子包括转座酶。在一些情况下,在扩增过程中使用机械剪切来使核酸片段化。在一些情况下,在扩增过程中添加核苷酸,可以通过添加其他蛋白质或条件使其片段化。例如,将尿嘧啶并入扩增子中;用尿嘧啶d-糖基化酶的处理使核酸在含尿嘧啶位置处片段化。在一些情况下,还采用了选择性核酸片段化的其他体系,例如,切割修饰的胞嘧啶-芘碱基对的工程化dna糖基化酶(kwon,等人chem biol.2003,10(4),351)。
[0135]
本文描述了包括使用终止子核苷酸的扩增方法,该终止子核苷酸终止核酸复制,从而减小扩增产物的大小。在一些情况下,这些终止子与本文所述的聚合酶、链置换因子或其他扩增组分结合使用。在一些情况下,终止子核苷酸减少或降低了核酸复制的效率。在一些情况下,这些终止子将延伸率减少至少99.9%、99%、98%、95%、90%、85%、80%、75%、70%或至少65%。在一些情况下,这些终止子将延伸率减少50%-90%、60%-80%、65%-90%、70%-85%、60%-90%、70%-99%、80%-99%或50%-80%。在一些情况下,终止子将平均扩增子产物长度减少至少99.9%、99%、98%、95%、90%、85%、80%、75%、70%或至少65%。在一些情况下,终止子将平均扩增子长度减少50%-90%、60%-80%、65%-90%、70%-85%、60%-90%、70%-99%、80%-99%或50%-80%。在一些情况下,包括终止子核苷酸的扩增子会形成环或发夹,从而降低聚合酶将这些扩增子用作模板的能力。在一些情况下,终止子的使用通过并入终止子核苷酸(例如,经过修饰以使其抵抗核酸外切酶从而终止dna延伸的双脱氧核苷酸)而减慢起始扩增位点处的扩增速度,从而生成较小的扩增产物。通过比当前使用的方法产生更小的扩增产物(例如,pta方法的平均长度为50-2000个核苷酸,而mda方法的平均产物长度为》10,000个核苷酸),在一些情况下pta扩增产物可直接进行连接衔接子而无需片段化,从而允许细胞条形码和唯一分子标识符(umi)的有效并入(参见图1h、图2b-图3e、图9、图10a和图10b)。
[0136]
终止子核苷酸以各种浓度存在,取决于诸如聚合酶、模板或其他因子等因素。例如,在一些情况下,在本文所述的方法中,终止子核苷酸的量表示为非终止子核苷酸与终止子核苷酸的比率。在一些情况下,这些浓度允许控制扩增子的长度。在一些情况下,终止子核苷酸与非终止子核苷酸的比率根据模板的存在量或模板的大小而改变。在一些情况下,对于较小的样品量,终止子核苷酸与非终止子核苷酸的比率会降低(例如,飞克到皮克的范围)。在一些情况下,非终止子核苷酸与终止子核苷酸的比率为约2:1、5:1、7:1、10:1、20:1、50:1、100:1、200:1、500:1、1000:1、2000:1或5000:1。在一些情况下,非终止子核苷酸与终止子核苷酸的比率为2:1-10:1、5:1-20:1、10:1-100:1、20:1-200:1、50:1-1000:1、50:1-500:1、75:1-150:1或100:1-500:1。在一些情况下,在使用本文所述的方法扩增期间存在的至少一个核苷酸是终止子核苷酸。每种终止子不必以约相同的浓度存在;在一些情况下,对于特定的一组反应条件、样品类型或聚合酶,可以优化本文所述方法中存在的各种终止子的比例。不受理论的约束,每种终止子在响应与模板链上相应核苷酸的配对时,并入扩增子的生长多核苷酸链中的效率可以不同。例如,在一些情况下,与胞嘧啶配对的终止子的浓度比平均终止子浓度高约3%、5%、10%、15%、20%、25%或50%。在一些情况下,与胸腺嘧啶配对的终止子的浓度比平均终止子浓度高约3%、5%、10%、15%、20%、25%或50%。在一些情况下,与鸟嘌呤配对的终止子的浓度比平均终止子浓度高约3%、5%、10%、15%、
20%、25%或50%。在一些情况下,与腺嘌呤配对的终止子的浓度比平均终止子浓度高约3%、5%、10%、15%、20%、25%或50%。在一些情况下,与尿嘧啶配对的终止子的浓度比平均终止子浓度高约3%、5%、10%、15%、20%、25%或50%。在一些情况下,能够终止通过核酸聚合酶的核酸延伸的任何核苷酸在本文所述的方法中用作终止子核苷酸。在一些情况下,可逆终止子用于终止核酸复制。在一些情况下,不可逆终止子用于终止核酸复制。在一些情况下,终止子的非限制性示例包括可逆和不可逆核酸和核酸类似物,例如,包括核苷酸的3’封闭的可逆终止子、包括核苷酸的3’未封闭的可逆终止子、包括脱氧核苷酸的2’修饰的终止子、包括对脱氧核苷酸的含氮碱基的修饰的终止子或其任何组合。在一个实施方案中,终止子核苷酸是双脱氧核苷酸。终止核酸复制并且可以适用于实施本发明的其他核苷酸修饰包括但不限于脱氧核糖的3’碳的r基团的任何修饰,如反向双脱氧核苷酸、3’生物素化核苷酸、3’氨基核苷酸、3
’‑
磷酸化核苷酸、3
’‑
o-甲基核苷酸、包括3’c3间隔子核苷酸、3’c18核苷酸、3’己二醇间隔子核苷酸的3’碳间隔子核苷酸、无环核苷酸,及其组合。在一些情况下,终止子是长度为1、2、3、4或更多个碱基的多核苷酸。在一些情况下,终止子不包括可检测的部分或标签(例如,质量标签、荧光标签、染料、放射性原子或其他可检测的部分)。在一些情况下,终止子不包括允许可检测部分或标签附接的化学部分(例如,“点击”叠氮化物/炔烃、共轭加成剂或用于标签附接的其他化学处理)。在一些情况下,所有终止子核苷酸都包括相同的修饰,该修饰减少核苷酸的某区域(例如,糖部分、碱基部分或磷酸部分)处的扩增。在一些情况下,至少一种终止子具有减少扩增的不同修饰。在一些情况下,所有终止子都具有基本相似的荧光激发或发射波长。在一些情况下,未修饰磷酸基团的终止子与不具有核酸外切酶校正活性的聚合酶一起使用。终止子在与具有可以除去终止子核苷酸的3
’‑
》5’校正核酸外切酶活性的聚合酶(例如,phi29)一起使用时,在一些情况下,还需要进一步修饰使其抵抗核酸外切酶。例如,双脱氧核苷酸被α-硫基修饰,产生硫代磷酸酯键,使这些核苷酸对核酸聚合酶的3
’‑
》5’校正核酸外切酶活性具有抗性。在一些情况下,这种修饰使聚合酶的核酸外切酶校正活性降低至少99.5%、99%、98%、95%、90%或至少85%。在一些情况下,提供对3
’‑
》5’核酸外切酶活性的抗性的其他终止子核苷酸修饰的非限制性示例包括:具有对α基团的修饰的核苷酸,如产生硫代磷酸酯键的α-硫代双脱氧核苷酸、c3间隔子核苷酸、锁核酸(lna)、反向核酸、2’氟碱基、3’磷酸化、2
’‑
o-甲基修饰(或其他2
’‑
o-烷基修饰)、丙炔修饰的碱基(例如,脱氧胞嘧啶、脱氧尿苷)、l-dna核苷酸、l-rna核苷酸、具有反向连接的核苷酸(例如,5
’‑5’
或3
’‑3’
)、5’反向碱基(例如,5’反向的2’,3
’‑
二脱氧dt)、甲基膦酸酯骨架和反式核酸。在一些情况下,具有修饰的核苷酸包括具有游离3’oh基团的碱基修饰的核酸(例如,2-硝基苄基烷基化的homedu三磷酸,具有大化学基团修饰如固体支持物或其他较大部分的碱基)。在一些情况下,将具有链置换活性但不具有3
’‑
》5’核酸外切酶校正活性的聚合酶与经历或未经历使其具有核酸外切酶抗性的修饰的终止子核苷酸一起使用。这些核酸聚合酶包括但不限于bst dna聚合酶、bsu dna聚合酶、deep vent(exo-)dna聚合酶、klenow片段(exo-)dna聚合酶、therminator dna聚合酶和ventr(exo-)。
[0137]
引物和扩增子文库
[0138]
本文描述了由至少一种靶核酸分子的扩增产生的扩增子文库。在一些情况下,这些文库是使用本文所述方法,如使用终止子的文库生成的。这些方法包括使用链置换聚合酶或因子、终止子核苷酸(可逆或不可逆)或本文所述的其他特征和实施方案。在一些情况
下,使用本文所述的终止子产生的扩增子文库在随后的扩增反应(例如,pcr)中进一步扩增。在一些情况下,随后的扩增反应不包括终止子。在一些情况下,扩增子文库包括多核苷酸,其中至少50%、60%、70%、80%、90%、95%或至少98%的多核苷酸包括至少一种终止子核苷酸。在一些情况下,扩增子文库包括衍生出扩增子文库的靶核酸分子。扩增子文库包括多种多核苷酸,其中至少一些多核苷酸是直接拷贝(例如,直接从靶核酸分子如基因组dna、rna或其他靶核酸复制)。例如,至少5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或超过95%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝。在一些情况下,至少5%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝。在一些情况下,至少10%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝。在一些情况下,至少15%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝。在一些情况下,至少20%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝。在一些情况下,至少50%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝。在一些情况下,3%-5%、3-10%、5%-10%、10%-20%、20%-30%、30%-40%、5%-30%、10%-50%或15%-75%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝。在一些情况下,至少一些多核苷酸是靶核酸分子的直接拷贝或子代(靶核酸的第一拷贝)。例如,至少5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或超过95%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝或子代。在一些情况下,至少5%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝或子代。在一些情况下,至少10%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝或子代。在一些情况下,至少20%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝或子代。在一些情况下,至少30%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝或子代。在一些情况下,3%-5%、3%-10%、5%-10%、10%-20%、20%-30%、30%-40%、5%-30%、10%-50%或15%-75%的扩增子多核苷酸是至少一种靶核酸分子的直接拷贝或子代。在一些情况下,靶核酸的直接拷贝的长度为50-2500、75-2000、50-2000、25-1000、50-1000、500-2000或50-2000个碱基。在一些情况下,子代的长度为1000-5000、2000-5000、1000-10,000、2000-5000、1500-5000、3000-7000或2000-7000个碱基。在一些情况下,pta扩增产物的平均长度为25-3000个核苷酸,为50-2500、75-2000、50-2000、25-1000、50-1000、500-2000或50-2000个碱基。在一些情况下,从pta产生的扩增子的长度不超过5000、4000、3000、2000、1700、1500、1200、1000、700、500或不超过300个碱基。在一些情况下,从pta产生的扩增子的长度为1000-5000、1000-3000、200-2000、200-4000、500-2000、750-2500或1000-2000个碱基。在一些情况下,使用本文所述方法产生的扩增子文库包括至少1000、2000、5000、10,000、100,000、200,000、500,000或超过500,000个包括唯一序列的扩增子。在一些情况下,文库包括至少100、200、300、400、500、600、700、800、900、1000、1100、1200、1300、1400、1500、2000、2500、3000或至少3500个扩增子。在一些情况下,长度小于1000个碱基的扩增子多核苷酸的至少5%、10%、15%、20%、25%、30%或超过30%是至少一种靶核酸分子的直接拷贝。在一些情况下,长度不超过2000个碱基的扩增子多核苷酸的至少5%、10%、15%、20%、25%、30%或超过30%是至少一种靶核酸分子的直接拷贝。在一些情况下,长度为3000-5000个碱基的扩增子多核苷酸的至少5%、10%、15%、20%、25%、30%或超过30%是至少一种靶核酸分子的直接拷贝。在一些情况下,直接拷贝扩增子与靶核酸分子的比例为至少10:1、100:1、1000:1、10,000:1、100,000:1、1,000,000:1、10,000,000:1或大于10,000,000:1。在一些情
况下,直接拷贝扩增子与靶核酸分子的比例为至少10:1、100:1、1000:1、10,000:1、100,000:1、1,000,000:1、10,000,000:1或大于10,000,000:1,其中直接拷贝扩增子的长度不超过700-1200个碱基。在一些情况下,直接拷贝扩增子和子扩增子与靶核酸分子的比例为至少10:1、100:1、1000:1、10,000:1、100,000:1、1,000,000:1、10,000,000:1或大于10,000,000:1。在一些情况下,直接拷贝扩增子和子扩增子与靶核酸分子的比例为至少10:1、100:1、1000:1、10,000:1、100,000:1、1,000,000:1、10,000,000:1或大于10,000,000:1,其中直接拷贝扩增子的长度为700-1200个碱基,并且子扩增子的长度为2500-6000个碱基。在一些情况下,文库包括约50-10,000、约50-5,000、约50-2500、约50-1000、约150-2000、约250-3000、约50-2000、约500-2000或约500-1500个扩增子,这些是靶核酸分子的直接拷贝。在一些情况下,文库包括约50-10,000、约50-5,000、约50-2500、约50-1000、约150-2000、约250-3000、约50-2000、约500-2000或约500-1500个扩增子,这些是靶核酸分子的直接拷贝或子扩增子。在一些情况下,直接拷贝的数量可以通过pcr扩增循环的次数来控制。在一些情况下,使用不超过30、25、20、15、13、11、10、9、8、7、6、5、4或3个pcr循环来生成靶核酸分子的拷贝。在一些情况下,使用约30、25、20、15、13、11、10、9、8、7、6、5、4或约3个pcr循环来生成靶核酸分子的拷贝。在一些情况下,使用3、4、5、6、7或8个pcr循环来生成靶核酸分子的拷贝。在一些情况下,使用2-4、2-5、2-7、2-8、2-10、2-15、3-5、3-10、3-15、4-10、4-15、5-10或5-15个pcr循环来生成靶核酸分子的拷贝。在一些情况下,使用本文所述方法生成的扩增子文库经受额外步骤,如衔接子连接和进一步的pcr扩增。在一些情况下,这些额外步骤在测序步骤之前。
[0139]
在一些情况下,由本文所述的pta方法和组合物(终止子、聚合酶等)生成的多核苷酸的扩增子文库具有增加的均匀性。在一些情况下,均匀性使用洛伦兹曲线(例如,图5c)或其他类似方法来描述。在一些情况下,这种增加使得覆盖所期望的靶核酸分子(例如,基因组dna、rna或其他靶核酸分子)所需的测序读取更少。例如,多核苷酸的累积分数的不超过50%包括靶核酸分子的序列的累积分数的至少80%的序列。在一些情况下,多核苷酸的累积分数的不超过50%包括靶核酸分子的序列的累积分数的至少60%的序列。在一些情况下,多核苷酸的累积分数的不超过50%包括靶核酸分子的序列的累积分数的至少70%的序列。在一些情况下,多核苷酸的累积分数的不超过50%包括靶核酸分子的序列的累积分数的至少90%的序列。在一些情况下,均匀性使用基尼指数描述(其中指数0表示文库的完全相等,指数1表示完全不等)。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.55、0.50、0.45、0.40或0.30。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.50。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.40。在一些情况下,这种均匀性度量取决于所获得的读取次数。例如,获得的读取不超过1亿、2亿、3亿、4亿或不超过5亿。在一些情况下,读取的长度为约50、75、100、125、150、175、200、225或约250个碱基。在一些情况下,均匀性度量取决于靶核酸的覆盖深度。例如,平均覆盖深度为约10x、15x、20x、25x或约30x。在一些情况下,平均覆盖深度为10-30x、20-50x、5-40x、20-60x、5-20x或10-20x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.55,其中获得了约3亿次读取。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.50,其中获得了约3亿次读取。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.45,其中获得了约3亿次读取。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.55,其中获得了不超过3亿
次读取。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.50,其中获得了不超过3亿次读取。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.45,其中获得了不超过3亿次读取。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.55,其中测序覆盖的平均深度为约15x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.50,其中测序覆盖的平均深度为约15x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.45,其中测序覆盖的平均深度为约15x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.55,其中测序覆盖的平均深度为至少15x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.50,其中测序覆盖的平均深度为至少15x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.45,其中测序覆盖的平均深度为至少15x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.55,其中测序覆盖的平均深度不超过15x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.50,其中测序覆盖的平均深度不超过15x。在一些情况下,本文所述的扩增子文库的基尼指数不超过0.45,其中测序覆盖的平均深度不超过15x。在一些情况下,使用本文所述方法生成的均匀扩增子文库需要经受额外步骤,如衔接子连接和进一步的pcr扩增。在一些情况下,这些额外步骤在测序步骤之前。
[0140]
引物包括用于引发本文所述的扩增反应的核酸。在一些情况下,这些引物包括但不限于具有或不具有使其抵抗核酸外切酶的修饰的任何长度的随机脱氧核苷酸,具有或不具有使其抵抗核酸外切酶的修饰的任意长度的随机核糖核苷酸,修饰的核酸,如锁核酸、靶向特定基因组区域的dna或rna引物,以及由诸如引发酶的酶引发的反应。在全基因组pta的情况下,优选使用具有随机或部分随机核苷酸序列的一组引物。在非常复杂的核酸样品中,不需要知道样品中存在的具体核酸序列,并且不需要将引物设计为与任何特定序列互补。相反,核酸样品的复杂性导致样品中大量不同的杂交靶序列,它们将与随机或部分随机序列的各种引物互补。在一些情况下,用于pta的引物的互补部分是完全随机的,仅包括随机的部分,或是选择性地随机的。在一些情况下,例如,引物的互补部分中的随机碱基位置数为引物的互补部分中的核苷酸总数的20%至100%。在一些情况下,引物的互补部分中的随机碱基位置数为引物的互补部分中的核苷酸总数的10%至90%、15-95%、20%-100%、30%-100%、50%-100%、75-100%或90-95%。在一些情况下,引物的互补部分中的随机碱基位置数为引物的互补部分中的核苷酸总数的至少10%、20%、30%、40%、50%、60%、70%、80%或至少90%。在一些情况下,使用标准技术合成具有随机或部分随机序列的引物组,这是通过允许在每个位置随机添加任何核苷酸。在一些情况下,引物组由具有相似的长度和/或杂交特性的引物组成。在一些情况下,术语“随机引物”是指在每个位置均可表现出4倍简并性的引物。在一些情况下,术语“随机引物”是指在每个位置均可表现出3倍简并性的引物。在一些情况下,本文所述方法中使用的随机引物包括长度为3、4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20或更多个碱基的随机序列。在一些情况下,引物包括长度为3-20、5-15、5-20、6-12或4-10个碱基的随机序列。引物也可以包括不可延伸的元件,其限制生成的扩增子的后续扩增。例如,在一些情况下,具有不可延伸元件的引物包括终止子。在一些情况下,引物包括终止子核苷酸,如1、2、3、4、5、10或超过10种终止子核苷酸。引物不需要局限于从外部添加至扩增反应的组分。在一些情况下,通过添加促进引发的核苷酸和蛋白质来原位生成引物。例如,在一些情况下,将与核苷酸组合的类似于引发酶的酶用于生成
本文所述方法的随机引物。在一些情况下,类似引发酶的酶是dnag或aep酶超家族的成员。在一些情况下,类似引发酶的酶是tthprimpol。在一些情况下,类似引发酶的酶是t7 gp4解旋酶-引发酶。在一些情况下,这些引发酶与本文所述的聚合酶或链置换因子一起使用。在一些情况下,引发酶用脱氧核糖核苷酸启动引发。在一些情况下,引发酶用核糖核苷酸启动引发。
[0141]
在pta扩增后可以选择扩增子的特定子集。在一些情况下,这种选择取决于大小、亲和力、活性、与探针的杂交或本领域中其他已知的选择因子。在一些情况下,选择在本文所述的额外步骤如衔接子连接和/或文库扩增之前或之后进行。在一些情况下,选择基于扩增子的大小(长度)。在一些情况下,选择较小的扩增子,其不太可能经历指数扩增,从而丰富了从原代模板衍生的产物,同时进一步将扩增从指数形式转化为拟线性扩增过程(图1a)。在一些情况下,选择长度为50-2000、25-5000、40-3000、50-1000、200-1000、300-1000、400-1000、400-600、600-2000或800-1000个碱基的扩增子。在一些情况下,通过使用方案例如在羧化顺磁珠上使用固相可逆固定化(spri)以富集特定大小的核酸片段或本领域技术人员已知的其他方案来进行大小选择。任选地或组合地,选择通过在制备测序文库时在pcr期间优先连接并扩增较小片段以及在测序(如合成测序、纳米孔测序或其他测序方法)过程中较小测序文库片段优先形成簇的结果来进行。选择较小片段的其他策略也符合本文所述的方法,并且包括但不限于在凝胶电泳后分离特定大小的核酸片段、使用结合特定大小的核酸片段的硅胶柱以及使用更强地富集较小片段的其他pcr策略。任何数量的文库制备方案都可以与本文所述的pta方法一起使用。在一些情况下,由pta生成的扩增子连接到衔接子上(任选去除终止子核苷酸)。在一些情况下,由pta生成的扩增子包括由基于转座酶的片段化生成的同源区域,这些区域被用作引发位点。在一些情况下,文库是通过机械方式或酶促方式对核酸进行片段化来制备的。在一些情况下,通过转座体的酶切法片段化(tagmentation)来制备文库。在一些情况下,通过连接衔接子,如y-衔接子、通用衔接子或环状衔接子来制备文库。
[0142]
pta中使用的引物的非互补部分可以包括可用于进一步操纵和/或分析扩增序列的序列。这种序列的一个示例是“检测标签”。检测标签具有与检测探针互补的序列,并使用其同源检测探针进行检测。引物上可以有一个、两个、三个、四个或超过四个的检测标签。除引物的大小外,对引物上可能存在的检测标签的数目没有基本限制。在一些情况下,引物上只有一个检测标签。在一些情况下,引物上有两个检测标签。当有多个检测标签时,它们可以具有相同的序列,也可以具有不同的序列,每个不同的序列与不同的检测探针互补。在一些情况下,多个检测标签具有相同的序列。在一些情况下,多个检测标签具有不同的序列。
[0143]
可以包含在引物的非互补部分中的序列的另一个示例是“地址标签”,其可以编码扩增子的其他细节,例如在组织切片中的位置。在一些情况下,细胞条形码包含地址标签。地址标签具有与地址探针互补的序列。地址标签被并入扩增链的末端。如果存在,引物上可以有一个或多个地址标签。除引物的大小外,引物上可能存在的地址标签的数目没有基本限制。当有多个地址标签时,它们可以具有相同的序列,也可以具有不同的序列,每个不同的序列都与不同的地址探针互补。地址标签部分可以是支持地址标签与地址探针之间特异性且稳定的杂交的任何长度。在一些情况下,来自多于一个来源的核酸可以并入可变标签序列。该标签序列的长度可以高达100个核苷酸,优选地长度为1至10个核苷酸,最优选地为
4、5或6个核苷酸,并且包括核苷酸的组合。在一些情况下,标签序列的长度为1-20、2-15、3-13、4-12、5-12或1-10个核苷酸。例如,如果选择六个碱基对形成标签并且使用四个不同核苷酸的排列,则可以制成总共4096个核酸锚(例如,发夹),每个锚具有唯一的6碱基标签。
[0144]
本文所述的引物可以存在于溶液中或固定在固体支持物上。在一些情况下,带有样品条形码和/或umi序列的引物可以固定在固体支持物上。例如,固体支持物可以是一个或多个珠子。在一些情况下,使个体细胞与一个或多个具有一组唯一的样品条形码和/或umi序列的珠子接触,以识别个体细胞。在一些情况下,将来自个体细胞的裂解物与一个或多个具有一组唯一的样品条形码和/或umi序列的珠子接触,以识别个体细胞裂解物。在一些情况下,将来自个体细胞的提取的核酸与一个或多个具有一组唯一的样品条形码和/或umi序列的珠子接触,以识别来自个体细胞的提取的核酸。珠子可以以本领域已知的任何合适方式来操纵,例如,使用本文所述的液滴致动器。珠子可以是任何合适的大小,包括例如,微珠、微粒、纳米珠和纳米颗粒。在一些实施方案中,珠子是磁响应的;在其他实施方案中,珠子没有明显的磁响应。合适的珠子的非限制性示例包括流式细胞术微珠、聚苯乙烯微粒和纳米颗粒、官能化的聚苯乙烯微粒和纳米颗粒、包覆的聚苯乙烯微粒和纳米颗粒、二氧化硅微珠、荧光微球和纳米球、官能化的荧光微球和纳米球、包覆的荧光微球和纳米球、颜色染色微粒和纳米颗粒、磁性微粒和纳米颗粒、超顺磁性微粒和纳米颗粒(例如,可从invitrogen group,carlsbad,ca获得的)、荧光微粒和纳米颗粒、包覆的磁性微粒和纳米颗粒、铁磁性微粒和纳米颗粒、包覆的铁磁性微粒和纳米颗粒,以及在美国专利申请公开号us20050260686、us20030132538、us20050118574、20050277197、20060159962中所描述的。珠子可以与抗体、蛋白质或抗原、dna/rna探针或任何其他对所需靶标具有亲和力的分子预耦合。在一些实施方案中,带有样品条形码和/或umi序列的引物可以在溶液中。在某些实施方案中,可以提供多个液滴,其中多个液滴中的每个液滴都具有对于液滴来说唯一的样品条形码和对于分子来说唯一的umi,从而使得umi在液滴集合内重复多次。在一些实施方案中,使个体细胞与具有一组唯一的样品条形码和/或umi序列的液滴接触,以识别个体细胞。在一些实施方案中,使来自个体细胞的裂解物与具有一组唯一的样品条形码和/或umi序列的液滴接触,以识别个体细胞裂解物。在一些实施方案中,将来自个体细胞的提取的核酸与具有一组唯一的样品条形码和/或umi序列的液滴接触,以识别从个体细胞中提取的核酸。各种微流控平台可用于分析单细胞。在一些情况下,通过流体动力学(液滴微流控、惯性微流控、涡流、微型阀、微观结构(如,微孔、微阱))、电学方法(介电泳(dep)、电渗透)、光学方法(光学镊子、光诱导介电泳(odep)、光热毛细管)、声学方法或磁性方法来操控细胞。在一些情况下,微流控平台包含微孔。在一些情况下,微流控平台包含基于pdms(聚二甲基硅氧烷)的装置。与本文所述方法兼容的单细胞分析平台的非限制性示例有:ddseq单细胞隔离器(bio-rad,hercules,ca,usa和illumina,san diego,ca,usa));chromium(10x genomics,pleasanton,ca,usa));rhapsody单细胞分析系统(bd,franklin lakes,nj,usa);tapestri平台(missionbio,san francisco,ca,usa));nadia innovate(dolomite bio,royston,uk);c1和polaris(fluidigm,south san francisco,ca,usa);icell8单细胞系统(takara);msnd(wafergen);puncher平台(vycap);cellraft air系统(cellmicrosystems);deparray nxt和deparray系统(menarini silicon biosystems);aviso cellcelector(als);indrop系统(1cellbio)和traptx(celldom)。
[0145]
pta引物可以包括序列特异性或随机引物、地址标签、细胞条形码和/或唯一分子标识符(umi)(参见,例如,图10a(线性引物)和图10b(发夹引物))。在一些情况下,引物包括序列特异性引物。在一些情况下,引物包括随机引物。在一些情况下,引物包括细胞条形码。在一些情况下,引物包括样品条形码。在一些情况下,引物包括唯一分子标识符。在一些情况下,引物包括两个或更多个细胞条形码。在一些情况下,这些条形码标识唯一的样品来源或唯一的工作流程。在一些情况下,这些条形码或umi的长度为5、6、7、8、9、10、11、12、15、20、25、30或超过30个碱基。在一些情况下,引物包括至少1000、10,000、50,000、100,000、250,000、500,000、106、107、108、109或至少10
10
个唯一条形码或umi。在一些情况下,引物包括至少8、16、96或384个唯一条形码或umi。在一些情况下,然后在测序前将标准衔接子连接至扩增产物上;测序后,首先根据细胞条形码将读取分配给特定细胞。可以与pta方法一起使用的合适衔接子包括,例如,可从integrated dna technologies(idt)获得的dual indexμmi衔接子。然后,使用umi将来自每个细胞的读取分组,并将具有相同umi的读取折叠为共有读取。使用细胞条形码允许在制备文库之前合并所有细胞,因为它们之后可以通过细胞条形码识别。在一些情况下,使用umi形成共有读取校正pcr偏倚,从而改善拷贝数变异(cnv)检测(图11a和图11b)。此外,可以通过要求来自同一分子的固定百分比的读取在每个位置具有相同的检测到的碱基变化来校正测序错误。这种方法已被用于改善cnv检测并校正大量样品中的测序错误。在一些情况下,umi与本文所述的方法一起使用,例如,美国专利号8,835,358公开了在附接随机可扩增条形码后的数字计数原理。schmitt.等人和fan等人(参见前文)公开了校正测序错误的类似方法。
[0146]
本文所述的方法还可以包括额外步骤,包括对样品或模板进行的步骤。在一些情况下,这些样品或模板在pta之前要经过一个或多个步骤。在一些情况下,对包括细胞的样品进行预处理步骤。例如,使用冻融、triton x-100、tween 20和蛋白酶k的组合对细胞进行裂解和蛋白水解,以增加染色质的可及性。其他裂解策略也适用于实施本文所述的方法。这些策略包括但不限于使用洗涤剂和/或溶菌酶和/或蛋白酶处理,以及/或细胞物理破坏如超声和/或碱裂解和/或低渗裂解的其他组合进行裂解。在一些情况下,用机械(例如,高压匀浆器、珠磨法)或非机械(物理方法、化学方法或生物方法)方式裂解细胞。在一些情况下,物理裂解方法包括加热、渗透压冲击和/或空蚀。在一些情况下,化学裂解包括碱和/或去垢剂。在一些情况下,生物裂解包括使用酶。裂解方法的组合也与本文所述的方法兼容。裂解酶的非限制性示例包括重组溶菌酶、丝氨酸蛋白酶和细菌裂解素。在一些情况下,用酶裂解包括使用溶菌酶、溶葡球菌酶、消解酶、纤维素酶、蛋白酶或聚糖酶。在一些情况下,对原代模板或靶分子进行预处理步骤。在一些情况下,使用氢氧化钠使原代模板(或靶标)变性,然后中和溶液。其他变性策略也可适用于实施本文所述的方法。这些策略可以包括但不限于将碱裂解与其他碱性溶液组合,提高样品温度和/或改变样品中的盐浓度,添加添加剂如溶剂或油,其他修饰或其任何组合。在一些情况下,额外步骤包括按大小对样品、模板或扩增子进行分类、过滤或分离。例如,在用本文所述的方法扩增后,扩增子文库富集具有期望长度的扩增子。在一些情况下,扩增子文库富含长度为50-2000、25-1000、50-1000、75-2000、100-3000、150-500、75-250、170-500、100-500或75-2000个碱基的扩增子。在一些情况下,扩增子文库富含长度不超过75、100、150、200、500、750、1000、2000、5000或不超过10,000个碱基的扩增子。在一些情况下,扩增子文库富含长度为至少25、50、75、100、150、200、500、
750、1000或至少2000个碱基的扩增子。
[0147]
本文所述的方法和组合物可包括缓冲剂或其他制剂。在一些情况下,这些缓冲剂包括表面活性剂/洗涤剂或变性剂(tween-20、dmso、dmf,包括疏水基团的聚乙二醇化聚合物或其他表面活性剂)、盐(磷酸钾或磷酸钠(一元或二元)、氯化钠、氯化钾)、trishcl、氯化镁或硫酸镁、如磷酸盐、硝酸盐或硫酸盐的铵盐、edta)、还原剂(dtt、thp、dte、β-巯基乙醇、tcep或其他还原剂)或其他组分(甘油、如peg的亲水性聚合物)。在一些情况下,将缓冲剂与诸如聚合酶、链置换因子、终止子或本文所述的其他反应组分等组分结合使用。缓冲剂可以包含一种或多种拥挤剂。在一些情况下,拥挤试剂包括聚合物。在一些情况下,拥挤试剂包括聚合物,例如多元醇。在一些情况下,拥挤试剂包括聚乙二醇聚合物(peg)。在一些情况下,拥挤试剂包含多糖。拥挤试剂的示例包括但不限于ficoll(例如,ficoll pm 400、ficoll pm 70或其他分子量的ficoll)、peg(例如,peg1000、peg2000、peg4000、peg6000、peg8000或其他分子量的peg)、葡聚糖(葡聚糖6、葡聚糖10、葡聚糖40、葡聚糖70、葡聚糖6000、葡聚糖138k或其他分子量的葡聚糖)。
[0148]
根据本文所述方法扩增的核酸分子可以使用本领域技术人员已知的方法进行测序和分析。在一些情况下,使用的测序方法的非限制性示例包括,例如,杂交测序(sbh)、连接测序(sbl)(shendure等人(2005)science 309:1728)、定量增量荧光核苷酸添加测序(qifnas)、逐步连接和切割、荧光共振能量转移(fret)、分子信标、taqman报告基因探针消化、焦磷酸测序、荧光原位测序(fisseq)、fisseq珠子(美国专利号7,425,431)、摆动测序(国际专利申请公开号wo2006/073504)、多重测序(美国专利申请公开号us2008/0269068;porreca等人,2007,nat.methods 4:931)、聚合酶克隆(polony)测序(美国专利号6,432,360、6,485,944和6,511,803,以及国际专利申请公开号wo2005/082098)、纳米网格滚环测序(rolony)(美国专利号9,624,538)、等位基因特异性寡核苷酸连接测定(例如,寡核苷酸连接测定(ola),使用连接的线性探针和滚环扩增(rca)读出的单模板分子ola,连接的挂锁探针和/或使用连接的圆形挂锁探针和滚环扩增(rca)读出的单模板分子ola)、高通量测序方法,例如,使用roche 454、illumina solexa、ab-solid、helicos、polonator平台等的方法,以及基于光的测序技术(landegren等人(1998)genome res.8:769-76;kwok(2000)pharmacogenomics1:95-100;以及shi(2001)clin.chem.47:164-172)。在一些情况下,将扩增的核酸分子进行鸟枪法测序。在一些情况下,使用任何适当的测序技术对测序文库进行测序,包括但不限于单分子实时(smrt)测序、聚合酶克隆测序、连接法测序、可逆终止子测序、质子探测测序、离子半导体测序、纳米孔测序、电子测序、焦磷酸测序、马克萨姆-吉尔伯特测序、链终止(如sanger)测序、+s测序或边合成边测序(基于阵列/集落或基于纳米球)。
[0149]
本文描述了使用本文所述的pta方法从包括短核酸的样品中生成扩增子文库的方法。在一些情况下,pta可以提高短核酸扩增的保真度和均匀性。在一些情况下,核酸的长度不超过2000个碱基。在一些情况下,核酸的长度不超过1000个碱基。在一些情况下,核酸的长度不超过500个碱基。在一些情况下,核酸的长度不超过200、400、750、1000、2000或5000个碱基。在一些情况下,包括短核酸片段的样品包括但不限于古dna(年龄为数百年、数千年、数百万甚至数十亿年)、ffpe(福尔马林固定石蜡包埋的)样品、无细胞dna或其他包括短核酸的样品。试剂盒
[0150]
本文描述了有助于实施pta方法的试剂盒。可以以试剂盒形式提供上文阐述的关于示例性反应混合物和反应方法的组分的各种组合。试剂盒可以包括彼此分开(例如,在不同的容器或包装中装载)的单独组分。在一些情况下,试剂盒包括本文所述组分的一种或多种子组合,该一种或多种子组合与试剂盒的其他组分分开。在一些情况下,这些子组合可组合成本文所述的反应混合物(或组合以进行本文所述的反应)。在特定的实施方案中,存在于单个容器或包装中的组分的子组合不足以进行本文所述的反应。然而,在一些情况下,试剂盒作为一个整体包括容器或包装的集合,其内容物可以组合以执行本文所述的反应。
[0151]
试剂盒可包括用于容纳该试剂盒的内容物的合适的包装材料。在一些情况下,包装材料由众所周知的方法制造,优选以提供无菌、无污染的环境。本文使用的包装材料包括,例如,那些通常用于与核酸测序系统一起使用的市售试剂盒中的包装材料。示例性的包装材料包括但不限于玻璃、塑料、纸、箔等,它们能够在固定限度内保持本文所述的组分。包装材料可以包括标签,该标签指示组分的特定用途。在一些情况下,标签所指示的试剂盒的用途是本文所述的一种或多种方法,其适合于试剂盒中存在的组分的特定组合。例如,在一些情况下,标签指示了该试剂盒可用于使用pta方法检测核酸样品中突变的方法。试剂盒中还可以包括包装的试剂或组分的使用说明书。说明书通常包括描述反应参数,例如试剂盒组分和待混合样品的相对量、试剂/样品混合物的维持时间段、温度、缓冲条件等的有形表述。应当理解,并非特定反应所需的所有组分都需要存在于特定试剂盒中。相反,在一些情况下,一种或多种附加组分由其他来源提供。在一些情况下,随试剂盒提供的说明书确定了待提供的附加组分以及从哪里可以获得这些组分。在一个实施方案中,试剂盒提供至少一种扩增引物;至少一种核酸聚合酶;至少两种核苷酸的混合物,其中核苷酸的混合物包括至少一种终止子核苷酸,该终止子核苷酸终止通过聚合酶的核酸复制;和该试剂盒的使用说明书。在一些情况下,该试剂盒提供执行本文所述方法的试剂,例如pta。在一些情况下,试剂盒还包括配置用于基因编辑(例如,crispr/cas9或本文描述的其他方法)的试剂。
[0152]
在相关方面,本发明提供了一种试剂盒,其包括逆转录酶、核酸聚合酶、一种或多种扩增引物、包含一种或多种终止子核苷酸的核苷酸混合物,以及任选的使用说明书。在本发明试剂盒的一个实施方案中,核酸聚合酶是链置换dna聚合酶。在本发明试剂盒的一个实施方案中,核酸聚合酶选自噬菌体phi29(φ29)聚合酶、基因修饰的phi29(φ29)dna聚合酶、dna聚合酶i的klenow片段、噬菌体m2 dna聚合酶、噬菌体phiprd1 dna聚合酶、bst dna聚合酶、bst大片段dna聚合酶、exo(-)bst聚合酶、exo(-)bca dna聚合酶、bsu dna聚合酶、vent
r dna聚合酶、ventr(exo-)dna聚合酶、deep vent dna聚合酶、deep vent(exo-)dna聚合酶、isopol dna聚合酶、dna聚合酶i、therminator dna聚合酶、t5 dna聚合酶、测序酶、t7 dna聚合酶、t7-测序酶和t4 dna聚合酶。在本发明试剂盒的一个实施方案中,核酸聚合酶具有3
’‑
》5’核酸外切酶活性,并且终止子核苷酸抑制这种3
’‑
》5’核酸外切酶活性(例如,带有α基团修饰的核苷酸[例如,α-硫代双脱氧核苷酸]、c3间隔子核苷酸、锁核酸(lna)、反向核酸、2’氟核苷酸、3’磷酸化核苷酸、2
’‑
o-甲基修饰的核苷酸、反式核酸)。在本发明试剂盒的一个实施方案中,核酸聚合酶不具有3
’‑
》5’核酸外切酶活性(例如,bst dna聚合酶、exo(-)bst聚合酶、exo(-)bca dna聚合酶、bsu dna聚合酶、ventr(exo-)dna聚合酶、deep vent(exo-)dna聚合酶、klenow片段(exo-)dna聚合酶、therminator dna聚合酶)。在一个特定的实施方案中,终止子核苷酸包括脱氧核糖的3’碳的r基团的修饰。在一个特定的实施方案
中,终止子核苷酸选自包括核苷酸的3’封闭的可逆终止子、包括核苷酸的3’未封闭的可逆终止子、包括脱氧核苷酸的2’修饰的终止子、包括对脱氧核苷酸的含氮碱基的修饰的终止子及其组合。在一个特定的实施方案中,终止子核苷酸选自双脱氧核苷酸、反向双脱氧核苷酸、3’生物素化核苷酸、3’氨基核苷酸、3
’‑
磷酸化核苷酸、3
’‑
o-甲基核苷酸、包括3’c3间隔子核苷酸、3’c18核苷酸、3’己二醇间隔子核苷酸的3’碳间隔子核苷酸、无环核苷酸及其组合。
[0153]
编号的实施方案
[0154]
本文描述了以下编号的实施方案1-104。1.本文提供了一种确定突变的方法,包括:a.将细胞群体暴露于基因编辑方法下,其中,该基因编辑方法利用被配置成在靶序列中实现突变的试剂;b.从该群体中分离出单细胞;c.提供来自单细胞的细胞裂解物;d.使细胞裂解物与至少一种扩增引物、至少一种核酸聚合酶和核苷酸混合物接触,其中核苷酸混合物包括至少一种终止子核苷酸,该终止子核苷酸终止通过聚合酶的核酸复制;和e.扩增靶核酸分子以生成多种终止的扩增产物,其中复制通过链置换复制来进行;f.将步骤(e)中获得的分子连接到衔接子上,从而生成扩增产物文库;g.对扩增产物文库进行测序;以及h.将扩增产物序列与至少一种参照序列进行比较,以识别至少一个突变。2.本文还提供了实施方案1的方法,其中至少一个突变存在于靶序列中。3.本文还提供了实施方案1的方法,其中至少一个突变不存在于靶序列中。4.本文还提供了实施方案1或2的方法,其中该基因编辑方法包括使用crispr、talen、zfn、重组酶或大范围核酸酶。5.本文还提供了实施方案1或2的方法,其中该基因编辑技术包括使用crispr。6.本文还提供了实施方案1或2的方法,其中该基因编辑技术包括使用基因治疗方法。7.本文还提供了实施方案6的方法,其中基因治疗方法未配置为修饰细胞的体细胞dna或种系dna。8.本文还提供了实施方案5的方法,其中,该参照序列是基因组。9.本文还提供了实施方案5的方法,其中,该参照序列是特异性决定序列,其中该特异性决定序列被配置为与靶序列结合。10.本文还提供了实施方案9的方法,其中至少一个突变存在于与特异性决定序列有至少1个碱基不同的序列区域中。11.本文还提供了实施方案9的方法,其中至少一个突变存在于与特异性决定序列有至少2个碱基不同的序列区域中。12.本文还提供了实施方案9的方法,其中至少一个突变存在于与特异性决定序列有至少3个碱基不同的序列区域中。13.本文还提供了实施方案9的方法,其中至少一个突变存在于与特异性决定序列有至少5个碱基不同的序列区域中。14.本文还提供了实施方案1的方法,其中该至少一个突变包含插入、缺失或置换。15.本文还提供了实施方案5的方法,其中该参照序列是crispr rna(crrna)序列。16.本文还提供了实施方案5的方法,其中该参照序列是单向导rna(sgrna)序列。17.本文还提供了实施方案5的方法,其中该至少一个突变存在于与催化活性cas9结合的序列区域中。18.本文还提供了实施方案1的方法,其中单细胞是哺乳动物细胞。19.本文还提供了实施方案1的方法,其中单细胞是人类细胞。20.本文还提供了实施方案1-19中任一项的方法,其中单细胞来源于肝脏、皮肤、肾脏、血液或肺。21.本文还提供了实施方案1-20中任一项的方法,其中单细胞是原代细胞。22.本文还提供了实施方案1-20中任一项的方法,其中单细胞是干细胞。23.本文还提供了实施方案1-20中任一项的方法,其中至少一些扩增产物包含条形码。24.本文还提供了实施方案1-20中任一项的方法,其中至少一些扩增产物包含至少两种条形码。25.本文还提供了实施方案23的方法,其中条形码包括细胞条形码。26.本文还提供了实施方案23或25的方法,其中条形
码包括样品条形码。27.本文还提供了实施方案1-26中任一项的方法,其中至少一些扩增引物包含唯一分子标识符(umi)。28.本文还提供了实施方案1-26中任一项的方法,其中至少一些扩增引物包含至少两种唯一分子标识符(umi)。29.本文还提供了实施方案1-27中任一项的方法,其中该方法还包括使用pcr的额外扩增步骤。30.本文还提供了实施方案1-29中任一项的方法,其中该方法还包括在连接到衔接子之前,从终止的扩增产物中去除至少一种终止子核苷酸。31.本文还提供了实施方案1-30中任一项的方法,其中使用包括微流控装置的方法从群体中分离出单细胞。32.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在少于50%的细胞群体中。33.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在少于25%的细胞群体中。34.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在少于1%的细胞群体中。35.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过0.1%的细胞群体中。36.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过0.01%的细胞群体中。37.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过0.001%的细胞群体中。38.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过0.0001%的细胞群体中。39.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过25%的扩增产物序列中。40.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过1%的扩增产物序列中。41.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过0.1%的扩增产物序列中。42.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过0.01%的扩增产物序列中。43.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过0.001%的扩增产物序列中。44.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变发生在不超过0.0001%的扩增产物序列中。45.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变存在于与遗传性疾病或病况相关的序列区域中。46.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变存在于与dna修复酶的结合不相关的序列区域中。47.本文还提供了实施方案1-31中任一项的方法,其中至少一个突变存在于与mre11的结合不相关的序列区域中。48.本文还提供了实施方案1-31中任一项的方法,其中该方法还包括识别先前由另一种脱靶检测方法测序的假阳性突变。49.本文还提供了实施方案48的方法,其中该脱靶检测方法是计算机模拟预测、chip-seq、guide-seq、circle-seq、htgts(高通量全基因组易位测序)、idlv(整合缺陷型慢病毒)、digenome-seq、fish(荧光原位杂交)或discover-seq。50.本文提供了识别特异性决定序列的方法,包括:a.提供核酸文库,其中至少一些核酸包含特异性决定序列;b.对至少一个细胞执行基因编辑方法,其中该基因编辑方法包括使细胞与包含至少一种特异性决定序列的试剂接触;c.使用实施方案1-38中任一项提供的方法对至少一个细胞的基因组进行测序,其中识别与至少一个细胞接触的特异性决定序列;和d.识别提供最少脱靶突变的至少一种特异性决定序列。51.本文还提供了实施方案50的方法,其中脱靶突变是沉默突变。52.本文还提供了实施方案50的方法,其中该脱靶突变存在于基因编码区之外。53.本文提供了体内突变分析的方法,包括:a.对活生物体中的至少一个细胞执行基因编辑方法,其中该基因编辑方法包括使细胞与包含至少一种特异性决定序列的试剂接触;b.从该生物体中分离出至少一个细胞;c.使用实施方案1-49中任一项提供的方法对至少一个细胞的基因组进行测序。54.本文
还提供了实施方案53的方法,其中该方法包括至少两个细胞。55.本文还提供了实施方案54的方法,其还包括通过比较第一细胞的基因组和第二细胞的基因组来识别突变。56.本文还提供了实施方案54或55的方法,其中第一细胞和第二细胞来自不同的组织。57.本文提供了预测对象年龄的方法,包括:a.提供来自对象的至少一个样品,其中该至少一个样品包含基因组;b.使用实施方案1-38中任一项提供的方法对基因组进行测序,以识别突变;c.将步骤b中获得的突变与标准参考曲线进行比较,其中该标准参考曲线将突变计数和位置与验证的年龄相关联;和d.基于与标准参考曲线的突变比较来预测对象的年龄。58.本文还提供了实施方案57的方法,其中该标准参考曲线针对对象的性别是特异性的。59.本文还提供了实施方案57的方法,其中该标准参考曲线针对对象的种族是特异性的。60.本文还提供了实施方案57的方法,其中该标准参考曲线针对对象的地理位置是特异性的,该对象在该地理位置度过了其生命中的一段时期。61.本文还提供了实施方案57-60中任一项的方法,其中该对象小于50岁。62.本文还提供了实施方案57-60中任一项的方法,其中该对象小于18岁。63.本文还提供了实施方案57-60中任一项的方法,其中该对象小于15岁。64.本文还提供了实施方案57-63中任一项的方法,其中该至少一个样品大于10年。65.本文还提供了实施方案57-63中任一项的方法,其中该至少一个样品大于100年。66.本文还提供了实施方案57-63中任一项的方法,其中该至少一个样品大于1000年。67.本文还提供了实施方案57-66中任一项的方法,其中对至少2个样品进行测序。68.本文还提供了实施方案57-66中任一项的方法,其中对至少5个样品进行测序。69.本文还提供了实施方案67的方法,其中该至少两个样品来自不同的组织。70.本文提供了对微生物或病毒基因组进行测序的方法,包括:a.获得包含一种或多种基因组或基因组片段的样品;b.使用实施方案1-38中任一项提供的方法对样品进行测序,以获得多个测序读取;和c.对测序读取进行组装和分选,以生成微生物或病毒基因组。71.本文还提供了实施方案70的方法,其中该样品包含来自至少两种生物体的基因组。72.本文还提供了实施方案70的方法,其中该样品包含来自至少十种生物体的基因组。73.本文还提供了实施方案70的方法,其中该样品包含来自至少100种生物体的基因组。74.本文还提供了实施方案70-73中任一项的方法,其中该样品来源为深海喷口、海洋、矿井、溪流、湖泊、陨石、冰川或火山的环境。75.本文还提供了实施方案70-74中任一项的方法,其还包括识别微生物基因组中的至少一种基因。76.本文还提供了实施方案70-75中任一项的方法,其中微生物基因组对应于未培养的生物体。77.本文还提供了实施方案76的方法,其中该微生物基因组对应于共生生物体。78.本文还提供了实施方案70-77中任一项的方法,其还包括在重组宿主生物体中克隆至少一种基因。79.本文还提供了实施方案78的方法,其中该重组宿主生物体是细菌。80.本文还提供了实施方案79的方法,其中该重组宿主生物体是埃希氏菌、芽孢杆菌或链霉菌。81.本文还提供了实施方案78的方法,其中该重组宿主生物体是真核细胞。82.本文还提供了实施方案81的方法,其中该重组宿主生物体是酵母细胞。83.本文还提供了实施方案82的方法,其中该重组宿主生物体是酵母或毕赤酵母。84.本文提供了用于核酸测序的试剂盒,包括:a.至少一种扩增引物;b.至少一种核酸聚合酶;c.至少两种核苷酸的混合物,其中核苷酸的混合物包括至少一种终止子核苷酸,该终止子核苷酸终止通过聚合酶的核酸复制;和d.使用试剂盒进行核酸测序的说明书。85.本文还提供了实施方案84的试剂盒,其中至少一种扩增引物是随机引物。86.本文还提供了实施方案84的试剂盒,其中该核酸聚合酶是dna聚合酶。87.本文还提供了实施方案86的试剂盒,其
中该dna聚合酶是链置换dna聚合酶。88.本文还提供了实施方案84-87中任一项的试剂盒,其中该核酸聚合酶是噬菌体phi29(φ29)聚合酶、基因修饰的phi29(φ29)dna聚合酶、dna聚合酶i的klenow片段、噬菌体m2 dna聚合酶、噬菌体phiprd1 dna聚合酶、bst dna聚合酶、bst大片段dna聚合酶、exo(-)bst聚合酶、exo(-)bca dna聚合酶、bsu dna聚合酶、ventr dna聚合酶、ventr(exo-)dna聚合酶、deep vent dna聚合酶、deep vent(exo-)dna聚合酶、isopol dna聚合酶、dna聚合酶i、therminator dna聚合酶、t5 dna聚合酶、测序酶、t7 dna聚合酶、t7-测序酶或t4 dna聚合酶。89.本文还提供了实施方案84-88中任一项的试剂盒,其中该核酸聚合酶包含3
’‑
》5’核酸外切酶活性,并且至少一种终止子核苷酸抑制该3
’‑
》5’核酸外切酶活性。90.本文还提供了实施方案84-88中任一项的试剂盒,其中该核酸聚合酶不包含3
’‑
》5’核酸外切酶活性。91.本文还提供了实施方案84-88中任一项的试剂盒,其中该聚合酶是bst dna聚合酶、exo(-)bst聚合酶、exo(-)bca dna聚合酶、bsu dna聚合酶、ventr(exo-)dna聚合酶、deep vent(exo-)dna聚合酶、klenow片段(exo-)dna聚合酶或therminator dna聚合酶。92.本文还提供了实施方案84-91中任一项的试剂盒,其中该至少一种终止子核苷酸包含脱氧核糖的3’碳的r基团的修饰。93.本文还提供了实施方案84-92中任一项的试剂盒,其中该至少一种终止子核苷酸选自于包含核苷酸的3’封闭的可逆终止子、包含核苷酸的3’未封闭的可逆终止子、包含脱氧核苷酸的2’修饰的终止子、包含对脱氧核苷酸的含氮碱基的修饰的终止子及其组合。94.本文还提供了实施方案84-93中任一项的试剂盒,其中该至少一种终止子核苷酸选自于双脱氧核苷酸、反向双脱氧核苷酸、3'生物素化核苷酸、3'氨基核苷酸、3'-磷酸化核苷酸、3'-o-甲基核苷酸、3'碳间隔子核苷酸(包括3'c3间隔子核苷酸)、3'c18核苷酸、3'己二醇间隔子核苷酸、无环核苷酸及其组合。95.本文还提供了实施方案84-94中任一项的试剂盒,其中该至少一种终止子核苷酸选自于含有对α基团的修饰的核苷酸、c3间隔子核苷酸、锁核酸(lna)、反向核酸、2'氟核苷酸、3'磷酸化核苷酸、2'-o-甲基修饰的核苷酸和反式核酸。96.本文还提供了实施方案84-95中任一项的试剂盒,其中含有对α基团的修饰的核苷酸是α-硫代双脱氧核苷酸。97.本文还提供了实施方案84-96中任一项的试剂盒,其中该扩增引物的长度为4至70个核苷酸。98.本文还提供了实施方案84-97中任一项的试剂盒,其中该至少一种扩增引物的长度为4至20个核苷酸。99.本文还提供了实施方案84-98中任一项的试剂盒,其中该至少一种扩增引物包含随机区域。100.本文还提供了实施方案99的试剂盒,其中该随机区域的长度为4至20个核苷酸。101.本文还提供了实施方案99或100的试剂盒,其中该随机区域的长度为8至15个核苷酸。102.本文还提供了实施方案84-101中任一项的试剂盒,其中该试剂盒还包括文库制备试剂盒。103.本文还提供了实施方案102的试剂盒,其中该文库制备试剂盒包含下列中的一种或多种:a.至少一种多核苷酸衔接子;b.至少一种高保真聚合酶;c.至少一种连接酶;d.用于核酸剪切的试剂;和e.至少一种引物;其中该引物被配置为与衔接子结合。104.本文还提供了实施方案84-103中任一项的试剂盒,其中该试剂盒还包含配置用于基因编辑的试剂。实施例
[0155]
提出以下实施例以更清楚地向本领域技术人员说明本文公开的实施方案的原理和实践,并且这些实施例不应被解释为限制任何要求保护的实施方案的范围。除非另有说明,否则所有份数和百分比均以重量计。
[0156]
实施例1:原代模板定向扩增(pta)
[0157]
尽管pta可以用于任何核酸扩增,但它对于全基因组扩增特别有用,因为与目前使用的方法如多重置换扩增(mda)相比,它允许以更均匀且可再现的方式捕获更大百分比的细胞基因组,并且错误率更低,避免了目前使用的方法的缺点,如在聚合酶首先延伸随机引物的位置处进行的指数扩增,而该指数扩增会导致基因座和等位基因的随机过度表达和突变传播(参见图1a-图1c)。
[0158]
细胞培养
[0159]
将人na12878(coriell institute)细胞维持在rpmi培养基中,该培养基补充有15%fbs和2mm的l-谷氨酰胺、100单位/ml的青霉素、100μg/ml的链霉素和0.25μg/ml的两性霉素b(gibco,life technologies)。细胞以3.5
×
105个细胞/ml的密度接种。培养物每3天分离一次,并在37c,5%co2的潮湿培养箱中维持。
[0160]
单细胞分离和wga
[0161]
以3.5
×
105细胞/ml的密度接种后,将na12878细胞培养至少三天,在这之后将3ml的细胞悬浮液以300xg沉淀10分钟。然后弃去培养基,并且用1ml的细胞洗涤缓冲剂(含有2%fbs,不含mg2或ca2的1x pbs)洗涤3次,以300xg、200xg和最后100xg离心5分钟。然后将细胞重悬于500μl细胞洗涤缓冲剂中。随后用100nm钙黄绿素am(分子探针)和100ng/ml碘化丙啶(pi;sigma-aldrich)染色,以区分活细胞群。将细胞加载至已被eliminase(decon labs)彻底清洗过的bd facscan流式细胞仪(facsaria ii)(bd biosciences)上,并使用accudrop荧光珠(bd biosciences)校准,以进行细胞分选。在要经历pta(sigma-aldrich)的细胞中将来自钙黄绿素am阳性且pi阴性的部分中的单细胞分入96孔板的每个孔中,孔中含有3μl的pbs(qiagen,repli-g sc试剂盒)和0.2%tween 20。有意将多个孔留空,以用作无模板对照(ntc)。分选后,立即将板短暂离心并置于冰上。然后,将细胞在-20℃冷冻至少过夜。第二天,在预pcr工作站上进行wga反应,该工作站提供恒定正压的hepa过滤空气,并在每次实验前用紫外线消毒30分钟。
[0162]
在进行mda时,采用了以前被证明可以提高扩增均一性的改进。具体而言,将抗核酸外切酶的随机引物添加到裂解缓冲剂/混合物中,达到125μm的终浓度。将4μl所得的裂解/变性混合物添加到含有单细胞的试管中,涡旋混合,短暂离心,并在冰上孵育10分钟。添加3μl淬灭缓冲剂来中和细胞裂解液,通过涡旋混合,短暂离心,并置于室温下。随后添加40μl扩增混合物,然后在30℃温育8小时,然后通过加热至65℃持续3分钟来终止扩增。
[0163]
pta是在冻融后首先进一步裂解细胞进行的,该裂解是通过添加2μl的5%triton x-100(sigma-aldrich)和20mg/ml蛋白酶k(promega)的1:1混合物的预冷溶液进行的。然后将细胞涡旋混合并短暂离心,之后在40℃放置10分钟。然后在裂解的细胞中添加4μl的裂解缓冲剂/混合液和1μl的500μm抗核酸外切酶的随机引物,以使dna变性,然后涡旋混合,离心,并在65℃放置15分钟。然后添加4μl室温淬灭缓冲剂,再将样品涡旋混合并离心。56μl扩增混合物(引物、dntp、聚合酶、缓冲剂),其在最终扩增反应中含有等比例的浓度为1200μm的α-硫代-ddntp。然后将样品在30℃放置8小时,之后将其加热至65℃持续3分钟来终止扩增。
[0164]
扩增步骤后,使用ampure xp磁珠(beckman coulter)以2:1的磁珠样品比纯化来自mda和pta反应的dna,并使用qubit dsdna hs测定试剂盒和qubit 3.0荧光计根据制造商的说明书(life technologies)来测量产量。
[0165]
文库制备
[0166]
mda反应产生的产量为40μg的扩增dna。按照标准程序,对1μg产物进行30分钟的酶促片段化。然后用15μm的双索引衔接子(通过t4聚合酶、t4多核苷酸激酶和taq聚合酶进行末端修复以加a尾)和4个pcr循环对样品进行标准文库制备。每个pta反应产生40-60ng用于标准dna测序文库的制备的材料。在用t4连接酶连接时使用了2.5μm带有umi和双索引的衔接子,并在最终扩增时使用了15个pcr循环(热启动聚合酶)。然后使用双侧spri来清理文库,右侧选择和左侧选择所采用的比例分别为0.65x和0.55x。在illumina nextseq平台上测序之前,使用qubit dsdna br测定试剂盒和2100生物分析仪(agilent technologies)对最终文库进行量化。包括novaseq在内的所有illumina测序平台也与该方案兼容。
[0167]
数据分析
[0168]
使用bcl2fastq基于细胞条形码对测序读取进行多路解编。然后使用trimmomatic修剪读取,随后使用bwa将其与hg19比对。读取由picard进行重复标记,然后使用gatk 4.0进行局部重新比对和碱基重新校准。所有用于计算质量指标的文件都使用picard downsamplesam下取样至2000万次读取。质量指标是使用qualimap以及picard alignmentsummarymetrics和collectwgsmetrics从最终bam文件获取的。总基因组覆盖也使用preseq估算。
[0169]
变异体判定
[0170]
使用来自gatk 4.0的gatk unifiedgenotyper判定单核苷酸变异体和插入/缺失。将使用gatk最佳实践的标准过滤条件用于过程中的所有步骤(https://software.broadinstitute.org/gatk/best-practices/)。使用control-freec(boeva等人,bioinformatics,2012,28(3):423-5)判定拷贝数变异体。结构变异体还使用crest检测(wang等人,nat methods,2011,8(8):652-4)。
[0171]
结果如图3a和图3b所示,仅用双脱氧核苷酸(“可逆”)扩增的映射率和映射质量得分分别为15.0+/-2.2和0.8+/-0.08,而掺入抗核酸外切酶的α-硫代双脱氧核苷酸终止子(“不可逆”)的映射率和映射质量得分分别为97.9+/-0.62和46.3+/-3.18。实验还使用可逆的ddntp和不同浓度的终止子进行(图2a,底部)。
[0172]
图2b-图2e示出了经过mda(按照dong,x.等人,nat methods.2017,14(5):491-493的方法)或pta的na12878人单细胞所产生的比较数据。虽然两种方案均产生了相当的低pcr重复率(mda为1.26%+/-0.52,而pta为1.84%+/-0.99)以及gc%(mda为42.0+/-1.47,而pta为40.33+/-0.45),但pta产生的扩增子更小。与mda相比,pta的映射读取百分比和映射质量得分也明显更高(分别为pta 97.9+/-0.62与mda 82.13+/-0.62,以及pta 46.3+/-3.18与mda 43.2+/-4.21)。总体而言,与mda相比,pta产生更多可用的映射数据。图4a示出,与mda相比,pta显著提高了扩增的均匀性,覆盖范围更广且覆盖接近0的区域更少。使用pta可以识别核酸群体中的低频序列变异体,包括占总序列的≥0.01%的变异体。pta可以成功用于单细胞基因组扩增。
[0173]
实施例2:pta的比较分析
[0174]
基准化pta和scmda细胞维持和分离
[0175]
将来自1000个基因组计划对象na12878(coriell institute,camden,nj,usa)的
类淋巴母细胞维持在rpmi培养基中,该培养基中补充了15%fbs、2mm的l-谷氨酰胺、100单位/ml的青霉素、100μg/ml的链霉素和0.25μg/ml的两性霉素b)。细胞以3.5
×
105细胞/ml的密度接种,并且每3天分离一次。将它们维持在37℃,5%co2的潮湿培养箱中。在单细胞分离前,将3ml在过去3天中已扩增的细胞的悬浮液以300xg离心10分钟。将沉淀的细胞用1ml细胞洗涤缓冲剂(含有2%fbs,不含mg
2+
或ca
2+
的1x pbs)洗涤3次,然后依次以300xg、200xg和最后100xg离心5分钟,以除去死细胞。然后将细胞重悬于500μl细胞洗涤缓冲剂中,然后用100nm钙黄绿素am和100ng/ml碘化丙啶(pi)染色,以区分活细胞群。将细胞加载至已被eliminase彻底清洗并使用accudrop荧光珠校准的bd facscan流式细胞仪(facsaria ii)上。将来自钙黄绿素am阳性且pi阴性部分的单细胞分入96孔板的每个孔中,孔中含有3μl的pbs和0.2%tween 20。有意将多个孔留空,以用作无模板对照。分选后,立即将板短暂离心并置于冰上。然后,将细胞在-80℃冷冻至少过夜。
[0176]
pta和scmda实验
[0177]
wga反应在预pcr工作站上组装,该工作站使用hepa过滤后的空气提供恒定正压,并在每次实验前用紫外线消毒30分钟。mda是根据scmda根据已公开的方案进行的(dong等人,nat.meth.2017,14,491-493)。具体而言,将抗核酸外切酶的随机引物以最终浓度12.5μm添加至裂解缓冲剂。将4μl所得的裂解混合物添加至含有单细胞的管中,移液3次以混合,短暂离心并在冰上温育10分钟。细胞裂解液通过添加3μl淬灭缓冲剂来中和,移液3次混合,短暂离心,并置于冰上。随后添加40μl扩增混合物,然后在30℃温育8小时,然后通过加热至65℃持续3分钟来终止扩增。pta通过在冻融后首先进一步裂解细胞来进行,该裂解是通过添加5%triton x-100和20mg/ml蛋白酶k的1:1混合物的2μl预冷溶液。然后将细胞涡旋并短暂离心,然后在40度放置10分钟。然后将4μl变性缓冲剂和1μl的500μm抗核酸外切酶的随机引物添加至裂解的细胞,以使dna变性,然后涡旋,离心并在65℃放置15分钟。然后4μl室温淬灭溶液,并将样品涡旋并离心分离。56μl扩增混合物含有等比例的在最终的扩增反应中浓度为1200μm的α-硫代-ddntp。然后将样品在30℃放置8小时,然后加热至65℃持续3分钟来终止扩增。在scmda或pta扩增后,使用ampure xp磁珠以2:1的磁珠样品比纯化dna,并使用qubit dsdna hs测定试剂盒和qubit 3.0荧光计根据制造商的说明书来测量产量。pta实验还使用可逆的ddntp和不同浓度的终止子进行(图2a,顶部)。
[0178]
文库制备
[0179]
根据标准方案,将1ug scmda产物酶促片段化30分钟。然后用15μm唯一双索引衔接子和4个pcr循环,对样品进行标准文库制备。在不进行片段化的情况下将每个pta反应的全部产物用于dna测序文库制备。在连接时使用2.5μm唯一双索引衔接子,并且在最终扩增中使用15个pcr循环。然后在1%琼脂糖e-gel上将来自scmda和pta的文库可视化。从凝胶切下400-700bp之间的片段,并使用gel dna回收试剂盒回收。在novaseq 6000上测序之前,使用qubit dsdna br测定试剂盒和agilent 2100生物分析仪对最终文库进行量化。
[0180]
数据分析
[0181]
使用trimmomatic修剪数据,随后使用bwa将其与hg19比对。读取由picard进行重复标记,然后使用gatk 3.5最佳实践进行局部重新比对和碱基重新校准。使用picard downsamplesam将所有文件下取样至指定的读取次数。质量指标是使用qualimap以及picard alignmentmetricsaummary和collectwgsmetrics从最终bam文件获取的。绘制洛伦
兹曲线,并使用htseqtools计算基尼指数。使用unifiedgenotyper进行snv判定,然后使用标准推荐条件(qd《2.0||fs》60.0||mq《40.0||sor》4.0||mqranksum《-12.5||readposranksum《-8.0)进行过滤。分析中没有排除任何区域,也没有进行其他数据标准化或操作。表1中列出了所测试方法的测序指标。
[0182]
表1:所测试的方法之间的测序指标的比较cv=变异系数;snv=单核苷酸变异;值是指15x覆盖。
[0183]
基因组覆盖范围和均匀性
[0184]
将pta与所有常见的单细胞wga方法进行综合比较。为实现此目的,分别对10个na12878细胞进行了pta和改进版本的mda,称为单细胞mda(dong等人.nat.meth.2017,14,491-493)(scmda)。此外,使用作为lianti研究的一部分产生的数据来比较通过dop-pcr(zhang等人,pnas 1992,89,5847-5851)、mda试剂盒1(dean等人,pnas 2002,99,5261-5266)、mda试剂盒2、malbac(zong等人,science 2012,338,1622-1626)、lianti(chen等人,science2017,356,189-194)或picoplex(langmore,pharmacogenomics 3,557-560(2002))扩增的细胞的结果。
[0185]
为了在样品之间进行标准化,将来自所有样品的原始数据进行排列并进行预处理,以使用相同的管线进行变异体判定。然后,在进行比较之前,将bam文件下取样至3亿次读取。重要的是,在进行进一步分析之前未筛选pta和scmda产物,而所有其他方法均在选择后续分析中使用的最高质量细胞之前进行了基因组覆盖和均匀性的筛选。值得注意的是,将scmda和pta与大量二倍体na12878样品进行比较,而所有其他方法均与lianti研究中使用的大量bj1二倍体成纤维细胞进行比较。如图3c-图3f所示,pta具有最高百分比的与基因组对齐的读取,以及最高的映射质量。pta、lianti和scmda具有相似的gc含量,均低于其他方法。在所有方法中,pcr复制率相似。此外,相对于其他经测试方法,pta方法使较小的模板(如线粒体基因组)能够提供更高的覆盖率(类似于较大的典型染色体)(图3g)。
[0186]
然后比较所有方法的覆盖范围和均匀性。示出了scmda和pta在染色体1上的覆盖图示例,其中示出,pta具有显著改善的覆盖均匀性和等位基因频率(图4b)。然后,使用增加
的读取次数来计算所有方法的覆盖率。pta在每个深度下都接近两种大量样品,这是相对于所有其他方法的显著改进(图5a)。然后,我们使用两种策略来衡量覆盖均匀性。第一种方法是在增加测序深度时计算覆盖变异系数,其中发现pta比所有其他方法更均匀(图5b)。第二种策略是计算每个下取样bam文件的洛伦兹曲线,其中再次发现pta具有最大的均匀性(图5c)。为了测量扩增均匀性的可再现性,计算了基尼指数以估算每个扩增反应与完全均匀性的差异(de bourcy等人,plos one 9,e105585(2014))。pta再次显示比其他方法可再现地更均匀(图5d)。
[0187]
snv灵敏度
[0188]
为了确定扩增方法性能的这些差异对snv判定的影响,在增加的测序深度下比较了每种方法与相应大量样品的变异体判定比率。为了估计灵敏度,比较了在每个测序深度下,在每个细胞中发现的在被下取样至6.5亿个读取的相应大量样品中判定的变异体百分比(图5e)。pta的覆盖和均匀性的改善使得检测出的变异体比mda试剂盒2多45.6%,而mda试剂盒2是第二高灵敏度的方法。在大量样品中判定为杂合的位点的检查显示,pta显著减少了那些杂合位点的等位基因倾斜(图5f)。这一发现支持了pta不仅在整个基因组中具有更均匀的扩增,而且在同一细胞中还更均匀地扩增两个等位基因的观点。
[0189]
snv精确性
[0190]
为了评估突变判定的精确性,在每个单细胞中判定的未在相应的大量样品中发现的变异体被认为是假阳性。scmda的较低温度裂解显著减少了假阳性变异体判定的数目(图5g)。使用热稳定聚合酶的方法(malbac、picoplex和dop-pcr)显示,随着测序深度的增加,snv判定的特异性进一步降低。不受理论约束,这很可能是这些聚合酶的错误率与phi29 dna聚合酶相比显著提高的结果。此外,在假阳性判定中看到的碱基改变模式也似乎是聚合酶依赖性的(图5h)。如图5g所示,与标准mda方案相比,pta中较低的假阳性snv判定率支持了pta中抑制错误传播的模型。此外,pta的假阳性变异体判定的等位基因频率最低,这再次与pta抑制错误传播的模型一致(图5i)。
[0191]
实施例3:环境致突变性的直接测量(dmem)
[0192]
pta被用于进行新型的致突变性测定,该测定为进行高分辨率、全基因组人类毒理基因组学研究提供框架。先前的研究,如ames测试,依靠细菌遗传学进行测量,这些测量被认为可代表人类细胞,但仅提供有关每个暴露细胞中诱导的突变数目和模式的有限信息。为了克服这些限制,开发了一种人类诱变系统“环境致突变性的直接测量(dmem)”,其中将单个人类细胞暴露于环境化合物中,分离为单细胞,并且进行单细胞测序,以识别在每个细胞中诱导的新突变。
[0193]
将表达干/祖细胞标志物cd34的脐带血细胞暴露于浓度增加的直接诱变剂n-乙基-n-亚硝基脲(enu)。已知enu具有相对较低的swain-scott底物常数,因此已被证明主要通过两步sn1机制起作用,该机制导致o4-胸腺嘧啶、o2-胸腺嘧啶和o2-胞嘧啶的优先烷基化。通过对靶基因的有限测序,enu在小鼠中也显示出对t到a(a到t)、t到c(a到g)和c到t(g到a)变化的偏好,这与在大肠杆菌中看到的模式有明显不同。
[0194]
用于致突变性实验的脐带血细胞的分离和扩增
[0195]
将enu(cas 759-73-9)和d-甘露醇(cas 69-65-8)以其最大溶解度放入溶液中。新鲜抗凝剂处理的脐带血(cb)从圣路易斯脐带血库获得。将cb用pbs以1:2稀释,并根据制造
商的说明书在ficoll-paque plus上通过密度梯度离心法分离单核细胞(mnc)。然后根据制造商的说明书,使用人cd34微珠试剂盒和磁性细胞分选(macs)系统对表达cd34的cb mnc进行免疫磁性选择。使用luna fl细胞计数仪测定细胞计数和活力。将cb cd34+细胞以2.5x104个细胞/ml的密度接种在补充了1x cd34+扩展补充剂、100单位/ml青霉素和100ug/ml链霉素的stemspan sfem中,在其中扩增96小时,之后进行诱变剂暴露。
[0196]
环境致突变性的直接测量(dmem)
[0197]
扩增的脐血cd34+细胞在补充有1x cd34+扩展补充剂、100单位/ml青霉素和100ug/ml链霉素的stemspan sfem中培养。将细胞暴露于浓度分别为8.54、85.4和854μm的enu、1152.8和11528μm的d-甘露醇或0.9%的氯化钠(媒介物对照)40小时。如上所述,收获来自药物处理的细胞和媒介物对照样品的单细胞悬浮液,并对其进行染色以测量活力。如上所述进行单细胞分类。按照本文所述方法和实施例2的一般方法,使用简化和改进的方案进行pta并制备文库。
[0198]
dmem数据的分析
[0199]
使用trimmomatic修剪从dmem实验中的细胞获取的数据,使用bwa将其与grch38比对,并使用gatk 4.0.1最佳实践进一步处理,未偏离推荐参数。使用haplotypecaller进行基因分型,再次使用标准参数过滤联合基因型。如果变异体的phred质量得分至少为100并且仅在一个细胞中发现,而在大量样品中却未发现,则仅被视为诱变剂的结果。通过使用bedtools从参照基因组中提取周围碱基,确定每个snv的三核苷酸背景。使用ggplot2和heatmap2对r中的突变计数和背景进行可视化。
[0200]
为了确定突变是否在cd34+细胞中的dna酶i超敏位点(dhs)中富集,计算了每个样品中与来自通过路线图表观基因组项目(roadmap epigenomics project)产生的10个cd34+原代细胞数据集的dhs位点重叠的snv的比例。dhs位点在两个方向都延伸了2个核小体或340个碱基。将每个dhs数据集与单细胞样品配对,在其中我们确定该细胞中与dhs重叠的至少10倍覆盖的人类基因组的比例,并将其与在覆盖的dhs位点中发现的snv的比例进行比较。
[0201]
结果
[0202]
与这些研究一致,观察到每个细胞的突变数目呈剂量依赖性增加,其中与媒介物对照或有毒剂量的甘露醇相比,在最低剂量的enu中检测到相似数目的突变(图12a)。也与先前使用enu的小鼠研究一致,最常见的突变是t到a(a到t)、t到c(a到g)和c到t(g到a)。还观察到其他三种类型的碱基变化,但c到g(g到c)的转化似乎是罕见的(图12b)。对snv的三核苷酸背景的检查示出了两种不同的模式(图12c)。第一种模式是当胞嘧啶后跟随鸟嘌呤时,胞嘧啶诱变似乎是罕见的。之后跟随着鸟嘌呤的胞嘧啶通常在人类基因组的第五个碳位点处被甲基化,这是异染色质的标志。不受理论的约束,假设由于异染色质的不可及性或由于5-甲基胞嘧啶与胞嘧啶相比的不利反应条件,5-甲基胞嘧啶不会被enu烷基化。为了检验前一假设,将突变位点的位置与cd34+细胞中已知的dna酶i超敏位点进行比较,这些位点由路线图表观基因组项目分类。如图12d所示,在dna酶i超敏位点中未观察到胞嘧啶变异体的富集。此外,在dh位点中未观察到限于胞嘧啶的变异体的富集(图12e)。此外,大多数胸腺嘧啶变异体发生在腺嘌呤位于胸腺嘧啶之前的地方。变异体的基因组特征注释与基因组中那些特征的注释没有显著差异(图12f)。
[0203]
实施例4:大规模并行单细胞dna测序
[0204]
使用pta,建立了用于大规模并行dna测序的方案。首先,将细胞条形码添加至随机引物。采用了两种策略来最大程度地减少细胞条形码引入的扩增中的任何偏差:1)延长随机引物的大小和/或2)创建引物,使其自身环回,以防止细胞条形码与模板结合(图10b)。一旦建立了最佳引物策略,则可以使用,例如,mosquito hts液体处理器对多达384种分选的细胞进行扩展,该处理器可以高精度地将甚至粘性液体移至25nl的体积。通过使用1μl pta反应代替标准的50μl反应体积,该液体处理器还可将试剂成本降低约50倍。
[0205]
通过将带有细胞条形码的引物递送至液滴,将扩增方案转化到液滴中。任选地使用固体支持物,如使用裂池策略创建的珠子。例如,合适的珠子可以从chemgenes获得。在一些情况下,寡核苷酸含有随机引物、细胞条形码、唯一分子标识符以及可切割的序列或间隔子,以在将珠和细胞封装在同一液滴中后释放寡核苷酸。在此过程中,优化液滴中低纳升体积的模板、引物、dntp、α-硫代-ddntp和聚合酶浓度。在一些情况下,优化包括使用较大的液滴以增加反应体积。如图9所示,此过程需要两个连续的反应来裂解细胞,然后进行wga。含有裂解细胞和珠子的第一液滴与含有扩增混合物的第二液滴结合。替代地或组合地,细胞在裂解前封装在水凝胶珠中,然后可以将两种珠子添加至油滴中。参见lan,f.等人,nature biotechnol.,2017,35:640-646)。
[0206]
其他的方法包括使用微孔,在一些情况下,该微孔捕获3
″×2″
的显微镜载玻片大小的装置上的20皮升反应室中的140,000个单细胞。与基于液滴的方法类似,这些孔将细胞与含有细胞条形码的珠子结合,从而允许进行大规模并行处理。参见gole等人,nature biotechnol.,2013,31:1126-1132。
[0207]
实施例5:pta在小儿急性成淋巴细胞白血病(all)中的应用
[0208]
已经对具有etv6-runx1易位的单个白血病细胞进行了单细胞外显子组测序,每个细胞测量了约200个编码突变,其中仅有25个存在于该患者的足够的细胞中并且可以通过标准大量测序检测到。然后,将每个细胞的突变负荷与这种类型的白血病的其他已知特征合并,如复制相关的突变率(1个编码突变/300次细胞分裂),从开始到诊断的时间(4.2年)以及诊断时的群体规模(1000亿个细胞),从而创建该疾病发展的计算机模拟。令人意外的是,即使在被认为是基因简单的癌症,如小儿all中,在患者诊断时,估计仍有3.3亿个克隆具有不同的编码突变谱。有趣的是,如图6b所示,通过标准大量测序仅检测到1至5个丰度最大的克隆(框c);有数千万个克隆由少量细胞组成,因此不太可能具有临床上显著的(框a)。因此,提供了用于增强检测灵敏度的方法,从而可以检测到构成细胞的至少0.01%(1:10,000)的克隆(框b),因为这是大多数可导致复发的抗药性疾病被假设所在的阶层。
[0209]
鉴于如此大量的种群遗传多样性,已经假设在给定的患者体内存在对治疗更有抵抗力的克隆。为了检验该假设,将样品置于培养物中,并将白血病细胞暴露于浓度不断增加的标准all化疗药物。如图7所示,在对照样品和接受最低剂量天冬酰胺酶的样品中,具有激活的kras突变的克隆继续扩展。然而,该克隆被证明对泼尼松龙和道诺霉素更敏感,而其他先前无法检测到的克隆在使用这些药物治疗后可以更清楚地被检测到(图7,虚线框)。这种方法还对处理过的样品进行了大量测序。在一些情况下,使用单细胞dna测序允许确定正在扩展的种群的多样性和克隆型。
[0210]
创建all克隆型药物敏感性的目录
[0211]
如图8所示,为了制作all克隆型药物敏感性的目录,取诊断样品的等分试样,并进行10,000个细胞的单细胞测序,以确定每种克隆型的丰度。同时,将诊断的白血病细胞在体外暴露于标准all药物(长春新碱、道诺霉素、巯嘌呤、泼尼松龙和天冬酰胺酶),以及一组靶向药物(依鲁替尼、达沙替尼和鲁索替尼)。选择活细胞,并且每次药物暴露至少对2500个细胞进行单细胞dna测序。最后,使用已建立的用于大量测序研究的方案,对来自完成6周治疗的相同患者的骨髓样品进行分选,以检测活的残留前白血病和白血病。然后,将pta用于以可扩展、高效且经济的方式对数万个细胞进行单细胞dna测序,从而实现以下目标。
[0212]
从克隆型到药物敏感性的药物敏感性目录一旦获得测序数据,就建立每个细胞的克隆型。为此,需要判定变异体并确定克隆型。通过利用pta,由目前使用的wga方法引入的等位基因缺失和覆盖偏倚受到限制。系统性地比较了用于从单细胞判定变异体的进行mda的工具,并且发现最近开发的工具monovar具有最高的敏感性和特异性(zafar等人,nature methods,2016,13:505-507)。一旦进行了变异体判定,就可以确定两个细胞是否具有相同的克隆型,尽管一些变异体判定由于等位基因缺失而丢失。为此,可以使用多元伯努利分布的混合模型(gawad等人,proc.natl.acad.sci.usa,2014,111(50):17947-52)。在确定细胞具有相同的克隆型后,确定要在目录中包括哪些变异体。包括满足以下任何条件的基因:1)它们是在大型儿科癌症基因组测序项目中发现的已知肿瘤抑制基因中出现的任何突变热点或功能丧失变异体(移码、无义、剪接)中检测到的非同义变异体;2)它们是在复发癌症样品中反复检测到的变异体;以及3)它们是在all患者接受了6周的治疗后在残留疾病的当前大量测序研究中得到阳性选择的复发变异体。如果克隆没有满足这些条件的至少两个变异体,则它们不包括在目录中。随着识别出更多与治疗抗性或疾病复发相关的基因,克隆可以被“救回”并包括在目录中。为了确定克隆型在对照与药物治疗之间经过阳性还是阴性选择,将费舍尔精确检验用于识别与对照有显著差异的克隆。仅当突变的至少两个一致的组合显示出与暴露于特定药物具有相同相关性时,才将克隆添加至目录。癌基因的已知激活突变或同一基因中肿瘤抑制基因的功能丧失突变被视为在克隆之间是等效的。如果克隆型不完全一致,则将共同的突变输入到目录中。例如,如果克隆型1是a+b+c,并且克隆型2是b+c+d,则b+c克隆型将输入到目录中。如果识别出在具有有限数目的同时发生的突变的抗性细胞中反复突变的基因,则这些克隆可以折叠成功能等效的克隆型。
[0213]
实施例6:测量单个人类细胞中crispr脱靶活性的比率和位置
[0214]
利用在单细胞中pta更高的变异体判定灵敏度和精确性,在单细胞中对高灵敏度的具有特定向导rna的crispr介导的基因组编辑进行了定量测量。对单细胞进行实施例4的常规pta方法。比较了未编辑的细胞和编辑后的细胞中的插入/缺失和sv计数(图13a和图13b)。
[0215]
还检查了这些基因组编辑方法可以在单个人类细胞中诱导的结构变异体的类型,结果如图14a-14c所示。如图14a所示,靶区域在底部(a)表示,并且位于6号染色体上的位置43,770,818和43,770,841之间(b)。以成对末端读取形式出现的序列数据(没有虚线的小水平条)表明单细胞序列数据和靶基因组(c)之间的一致性。读取内的虚线表示相对于参照基因组的基因组缺失(d)。在本实施例中,两个编辑后的细胞都显示与靶位点(a)重叠的缺失(d)。相反,两个未编辑的细胞包含表明它们与这个位置处的参照基因组一致的读取,因此
没有发生编辑。图14b示出了检测crispr诱导的编辑产生的大量(》1kb)缺失,该缺失仅限于编辑后的#1细胞。靶区域在底部(a)表示,并且位于18号染色体上的位置23,779,588和23,779,611之间(b)。读取形式的序列数据(彩色的小水平条,通常为灰色)表明单细胞序列数据和靶基因组(c)之间的一致性。比对读取突然下降的区域表明在这些位置处偏离了参照基因组。在这种情况下,18号染色体上位置23,778,472和23,779,607之间的读取覆盖的突然丢失表明编辑后的#1细胞中存在大量缺失(d)。这种缺失被确定为crispr诱导的缺失,因为图中最右侧的断点与基因组中和靶位点(a)高度相似的区域重叠,并且在未编辑的细胞中不存在这种缺失。(a)中的小写字母表示与靶位点不同的碱基。图14c示出了在编辑后的#1细胞中检测2号染色体位置241,275,213和4号染色体位置38,536,006之间的染色体间易位。易位断点与每条染色体中的grna脱靶区域重叠,这些区域与grna靶位点相似,并在底部[(a)和(b)]表示。左图表示与含有断点的2号染色体区域比对的读取,而右图表示与含有断点的4号染色体区域比对的读取。编辑后的#1细胞分为两个视图:与断点周围区域比对的所有读取的视图(c),和相同区域但仅显示作为易位证据的读取对(d)的视图。对于支持易位的读取对,一对中的一个读取与2号染色体比对,在断点处覆盖突然下降,另一个读取与4号染色体比对,在断点处读取覆盖也突然下降(e)。这种易位被识别为crispr诱导的易位,因为至少一个易位断点与基因组中和编辑后的细胞中靶位点高度相似的区域重叠(在这种情况下有两个:a和b),而在未编辑的细胞中没有易位的证据。(a)和(b)中的小写字母表示与靶位点不同的碱基。
[0216]
为了确认假定的脱靶位点,以及评估随着向导rna基因组错配数量的增加而产生的变异体判定的精确性,还对所有细胞中假定的脱靶位点进行了基于微流控高通量pcr的再测序(数据未显示)。
[0217]
实施例7:年龄评估
[0218]
收集了至少1000名人类对象的数据,包括地理位置(度过大部分时间的位置)、性别、年龄、种族以及利用pta方法建立的基因组突变频率和位置。样本一式两份,并从每个对象的一种或多种组织中获取样品。生成标准曲线,将诸如地理位置(居住时间最长的地区)、性别、年龄、种族、突变频率、突变位置或获得的其他数据的变量与对象的年龄相关联。使用pta方法对来自未知年龄对象的样品的基因组进行测序,并使用标准曲线来确定个体的年龄。如果已知关于该对象的其他信息(种族、地理位置),这将用于进一步改进预测。
[0219]
实施例8:临床细菌样品的鉴定和诊断
[0220]
获得来自患有疑似细菌感染的对象的细胞样品,并使用pta方法进行单细胞基因组测序。用pta方法确定的突变与赋予已知抗生素抗性的突变进行比较,或用于鉴定细菌菌株。使用这些信息来选择适当的治疗方法,诸如有效的抗生素。
[0221]
实施例9:微生物物种和基因的鉴定
[0222]
从各种来源诸如深海喷口、海洋、矿井、溪流、湖泊、陨石、冰川或火山采集水样。使样品经过20微米预过滤器以去除微粒,然后分级为诸如3-20微米、0.8-3微米、0.1-0.8微米和50kda至0.1微米的粒径组。然后对样品进行处理以分离出单个细胞,或者任选地进行批量处理。使用标准技术分离出基因组、质粒或其他dna,经pta方法处理,然后进行测序。基因组序列重新组装后,鉴定已知物种,并对未知物种和/或基因进行表征,以用于潜在的工业应用。
[0223]
实施例10.测量基因治疗方法的非预期插入率
[0224]
利用在单细胞中pta提高的变异体判定灵敏度和精确性,对单细胞中高灵敏度的基因治疗方法的非预期插入率进行定量测量。该方法可以通过检测周围序列来检测特定序列在非期望位置的插入,以确定基因治疗方法是否导致宿主基因组的插入或修饰。将编码产生蛋白质的基因的核酸导入病毒载体,然后递送至生物体内或体外的一种或多种细胞。病毒将核酸递送至细胞核,然后核酸被转录成mrna。mrna翻译后,产生蛋白质。使用实施例4中描述的常规pta方法对通过该基因治疗方法修饰的细胞进行测序,并检测由该基因治疗方法引起的突变(突变频率和位置/模式)。
[0225]
实施例11.在原代癌细胞中用pta判定cnv
[0226]
与mda以及最近开发或改进的市售试剂盒相比,按照实施例1的常规方法,使用原代白血病细胞对snv和拷贝数变异(cnv)判定的pta方案进行进一步的验证研究,该pta方案显示了覆盖范围的进一步提高,并且仍然是基于在碱基对分辨率处的cv计算的最均一的方法(图19)。在所有测序深度,pta还仍然是最灵敏的snv判定方法,并且目前通过改变为低温裂解还具有最高的snv判定特异性。依赖pcr的方法(wga试剂盒3,picoplex gold)随着测序深度的增加也仍然显示出特异性降低,尽管该特异性的降低比malbac和以前版本的picoplex有显著改善。
[0227]
为了评估每种方法判定不同大小cnv的准确性,将每个bam文件取样至3亿次读取,并在不断增大的面元(bin)大小下测量cv(图5j)。在每个面元下,发现与所有其他wga方法相比,pta具有最低的cv(图5j)。随着深度的增加,wga试剂盒2和picoplex gold的cv值急剧下降。这种特定的白血病样品在5q和11q上具有已知的cnv。正如预期的那样,大量样品和单细胞都检测到了x染色体的单个拷贝。cnv分析发现5q缺失是克隆性的,而11q改变仅在细胞的亚群内发现(图5k,阴影箭头)。批量数据表明在12p上可能有缺失,但是它在大量样品中未被判定。发现五个单细胞中的两个在相同的位置具有cnv,这表明单细胞cnv分析可能更灵敏,也是用于评估组织中具有给定拷贝数变化的细胞的百分比的更好策略。
[0228]
实施例12.同源细胞中snv率的测量
[0229]
通过将单个cd34+cb细胞接种到单孔中,随后扩增5天,进行同源细胞研究(图16a)。然后从该培养物中再分离出单细胞,以比较基因上几乎完全相同的细胞的变异体判定情况。此外,以大量细胞为参照,对种系、假阳性和体细胞变异体判定进行了区分(图16b)。利用这种方法,并再次使用大量样品作为实况,使用gatk4联合基因分型的低温方案确定的变异体判定精确性提高到99.9%(图16c)。此外,这些原代细胞中的大多数具有类似或改进的变异体检测灵敏度。然而,有一个细胞的变异体判定灵敏度显著降低,在不受理论束缚的情况下,这可能是人工操控脆性原代细胞的结果。另外,两个具有较高变异体判定灵敏度的细胞具有较少的纯合体细胞变异体判定,这可能是等位基因脱落减少的结果(图15b)。这些细胞中假阳性变异体倾向于较低的等位基因频率,在不受理论束缚的情况下,这可以解释为这些快速分裂的细胞在细胞周期的晚s或g2/m期时为四倍体,其中四个等位基因中只有一个获得复制错误(图17a-17c)。观察到纯合假阳性判定聚集在特定位置,而杂合判定则没有。在不受理论束缚的情况下,这可能是在扩增过程中位于这些位置的一个等位基因丢失或缺乏模板变性的结果,这似乎并不依赖于基因组区域的gc含量(图18a-18c)。大多数假阳性和体细胞变异体被判定为杂合子,这与分别因复制错误或在发育过程中导致的
只有一个等位基因发生突变的模型相一致(图16d)。在新生儿cd34+造血细胞中测量假阳性和体细胞突变率,被分别评估为每mb基因组0.9和1.4个。
[0230]
实施例13:测量单个人类细胞中crispr脱靶活性的比率和位置
[0231]
从修正导致或促成疾病形成的基因到根除目前无法治愈的感染性疾病,基因组编辑工具的持续发展显示出改善人类健康的巨大前景。然而,这些干预的安全性仍然不清楚,因为还不完全了解这些工具如何与编辑细胞基因组中的其他位置相互作用并永久改变这些位置。已经开发了评估基因组编辑策略的脱靶率的方法,但迄今为止已开发的所有工具都是一起质询(interrogate)细胞群,限制了测量每个细胞脱靶率和细胞间差异的能力,以及检测发生在少数细胞中的罕见编辑事件的能力。已经对编辑后的细胞进行了单细胞克隆,但是可以选择获得致命脱靶编辑事件的细胞,而且对许多类型的原代细胞来说是不切实际的。
[0232]
利用pta的提高的变异体判定灵敏度和特异性,获得了在单细胞中具有特异性向导rna(grna)的crispr介导的基因组编辑的定量测量(图20a)。这些研究使用了三种细胞类型:u20s骨肉瘤细胞系、原代造血cd34+cb细胞和胚胎干(es)细胞。此外,还使用了两种先前描述的grna,一种已知是精确的grna(emx1),另一种已知具有高水平的脱靶活性(vegfa)。为了确定具有高特异性的插入/缺失,变异体判定仅限于与pam序列完全匹配并与前间隔序列存在最多5个错配的基因组位置(图16a)。
[0233]
与单独接受cas9或进行模拟转染的对照细胞相比,在vegfa编辑后的细胞中有更多的脱靶插入/缺失,显示出广泛的细胞间差异,同时仅检测到少量脱靶emx1编辑事件(图20b)。我们注意到,在对照细胞中看到的大多数假定的假阳性编辑是单碱基对插入。去除非反复出现的的单碱基对插入进一步提高了插入/缺失判定的特异性(图21)。大多数(但并非所有)反复出现的脱靶位点都是细胞类型特异性的,进一步支持了这样的发现,即细胞类型的常规染色质结构影响脱靶基因组位置(图20d)。进行结构变异体(sv)判定以确定基因组编辑诱导的sv,其中要求两个断点周围的区域与pam序列完全匹配,并允许与前间隔序列存在最多5个错配。测量到使用vegfa向导rna的sv数量增加,在emx1编辑后的细胞中检测到仅1个sv,而在对照细胞中未检测到sv(图20e)。检测到反复出现的vegfa介导的sv,其中一些是细胞类型特异性的,并且在es细胞中检测到更大的sv(图20c)。
[0234]
实施例14:用pta组装细菌基因组
[0235]
获取口腔拭子并在lb培养基中培养过夜。将单个菌落分入96孔板中作为单独样品,并对每个孔执行实施例1的常规pta方法,以制备用于测序的每个样品。每个样品获得100-200万个读取,并使用spades(基于重叠群的方法)组装读取。图22a显示了10种不同细菌样品的最长重叠群数据。对于测序数据的计算机综合分析,按照长度递减的顺序依次添加每个样品的重叠群(图22b)。细菌样品10的数据显示在图22c中。然后,确定分配给每个属的总组装比例。污染物序列与基因组dna的小片段一起出现;这些可以被识别为数据集中较小的重叠群(》5kb,图22d)。如果两个读取都与grch38-重叠群联合参照中的grch38对齐,则认为读取对来自人类(图22e-22f)。备选地,采用来自参考数据库的k-mers将读取分配给分类群,对所有样品均使用免组装方法(例如kraken)。对细菌样品10的基于读取的方法的结果的显示于图22g,并与基于重叠群的方法一致。
[0236]
实施例15:用pta进行植入前的基因测试
[0237]
根据kuznyetsov等人(2018)plos one,13(5):e0197262的常规方法,通过制备20个培养的胚胎(冷冻的或新鲜的)进行非侵入性植入前遗传筛查(nipgs)。简言之,在培养的第4天,将每个胚胎转移到含有hsa的新鲜global hp培养基中,并在油下培养,直到达到囊胚期(在第5天或第6天)。达到完全扩张的囊胚后,每个囊胚都要接受激光辅助滋养外胚层活检,然后进行激光折叠,从而使bf与bccm混合。然后将胚胎转移到低温保存培养基中,并通过玻璃化来冷冻。取出胚胎后,收集合并的bccm和bf样品,并在-80℃下冷冻,直至测试。从bccm/bf样品中提取核酸后,对该核酸执行实施例1的常规pta方法。然后分析由pta生成的基因组dna文库的基因突变,如染色体异常。
[0238]
尽管本文中已经示出并描述了本发明的优选实施方案,但对于本领域技术人员显而易见的是,这些实施方案仅以示例的方式提供。本领域技术人员在不脱离本发明的情况下现将想到多种变化、改变和替代。应当理解,本文中所述的本发明实施方案的各种替代方案可用于实施本发明。以下权利要求旨在限定本发明的范围,并由此涵盖这些权利要求范围内的方法和结构及其等同项。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1