新颖金属氢化物的合成和储氢特性的制作方法
技术领域
本发明涉及用于制备金属氢化物的改进方法。本发明还涉及金属氢化物,例如本文所述方法所制备的金属氢化物,其用作储氢系统时,展现增强的储氢能力。
背景技术:
对世界化石燃料储备的巨大需求已经导致关于全球变暖、能量安全和环境污染的问题。研究人员继续寻找替代燃料来源。分子氢因为轻质、丰裕,质量能量密度超过当前所用烃类燃料(如汽油)的三倍,且其仅有的燃烧产物(水)对环境无害,所以就此而言,分子氢是理想的。尽管燃料电池技术已获得进展,但氢制备、储存仍然是个大障碍。参见例如R.H.Wiswall等人,《科学(Science)》,186,1158,1974;S.Orimo等人,《化学评论(Chem.Rev.)》,107,4111,2007;以及L.K.Heung,《使用金属氢化物的机载储氢系统(On-board Hydrogen Storage System Using Metal Hydride)》,《假设II(HYPOTHESIS II)》,1,1997。使用当前技术储氢的体积储能密度低于烃类燃料。因此,在所有其它因素均等的情况下,为了储存相同量的能量,储氢需要的储槽比储存烃燃料大得多且重得多。
重量法测定容量是一种每单位质量储存系统能够储存的氢量的度量。体积法测定容量是每单位容积储存系统能够储存的氢量的度量。美国能源部(The United States Department of Energy;DOE)已经设定储氢的目标。对于接近室温操作的完全可逆系统来说,DOE设定的2017年储氢目标是5.5wt%和40kg/m3的体积吸收能力。最终目标是7.5wt%和70kg/m3。
迄今为止,尚无满足DOE所提出的所有要求的技术。正在考虑的一些技术涉及使用化学载体,如合金;吸附剂,如无定形碳(参见例如R.Yang等人,《美国化学学会杂志(J.Am.Chem.Soc.)》,131,4224,2009)、沸石(参见例如A.等人,《物理化学杂志C(J.Phys.Chem.C)》,112,2764,2008)以及金属有机架构(metal organic frameworks;MOF)(参见例如K.M.Thomas,《道尔顿学报(Dalton Trans.)》,1487,2009;S.S.Kaye等人,《美国化学学会杂志》,129,14176,2007;以及N.L.Rosi等人,《科学》,300,1127,2003)。
热管理问题以及缓慢动力学和/或可逆性问题阻碍了金属氢化物(如LiH和NaAlH4)的使用。举例来说,当氢与镁或钠铝合金反应而得到金属氢化物(如MgH2和NaAlH4)时,放出大量的热。产生此热时,必须执行冷却步骤以防止系统温度显著上升,并且此冷却步骤对于系统来说构成了能量损失。另外,典型地需要加热以在需要时除去氢。这是氢化物(如MgH2和NaAlH4)典型的氢结合焓高(>60kJ/mol)的假象。
压缩技术已经用于增加气体压力和提高氢的体积储能密度。由此可以使储槽更小。然而,压缩氢需要大量的能量,通常占所储存能量的多达30%。另外,这种压缩技术需要大型压力容器。
另一种储氢技术涉及将氢气转变为液氢。因为氢的沸点极低(-252.88℃),所以这种技术需要低温储存。氢液化需要大量能量来维持这些极低温度。另外,液氢储槽需要复杂且昂贵的隔热以防止液氢蒸发。另外,液氢的体积能量密度低于烃类燃料(如汽油),是其约4分之一。
物理吸附材料,如无定形碳和金属有机架构(MOF),在77K温度下实现有前景的储存能力,但其性能在室温下由于吸收热较低(典型地是5-13kJ/mol H2)而损失约90%。参见例如A.Dailly等人,《物理化学杂志B(J.Phys.Chem.B)》,110,1099,2006;J.Rowsell等人,《应用化学国际版(Angew.Chem.,Int.Ed.)》,2005,4670,2005。为了在环境条件下实现DOE目标,预计理想的H2结合能是在每个氢分子20-30kJ/mol范围内。参见例如R.Lochan等人,《物理化学与化学物理(Phys.Chem.Chem.Phys.)》,8,1357,2006。此外,用于制备储氢材料的能量制备成本可以是重要因素。
因此,需要可以用作储氢系统的经改进的、成本较低的材料。另外,需要合成较高纯度材料的改进方法,所述材料当用作储氢系统时展现增强的储氢能力。
技术实现要素:
在一个方面中,本发明人已惊人地开发出一种制备适用于储氢应用中的金属氢化物化合物的改进方法。改进方法涉及使金属烃类化合物(例如金属烷基或金属芳基化合物)在氢不存在下发生热沉淀,随后使所得沉淀物发生氢化。本发明人已惊人地发现热沉淀方法形成了含有中间物的残余烃,不希望受理论所束缚,人们认为这是桥接模式。再次不希望受理论所束缚,本发明人推测出,沉淀方法可能通过可形成桥接亚烷基结构的α-消去(例如在双[(三甲基硅烷基)甲基]化合物的情况下,为四甲基硅烷的α-消去),或在金属芳基化合物的情况下通过双分子C-H活化的缩合和随后烃消去(即,双分子σ键复分解)来形成聚合物。人们相信,这些桥接配位体在下游无定形结构中产生空间,从而有效地充当模板以确保一旦桥接烃除去,则氢可以扩散于所述结构中以及离开所述结构。沉淀物氢化,随后除去残余烃。再次不希望受理论所束缚,本发明人推测所得金属氢化物含有桥接氢化物配位体。
本文所述的金属氢化物,如通过本文所述方法制备的那些金属氢化物,惊人地展现增强的储氢能力且允许金属中心与多个H2分子形成相互作用(例如库巴斯相互作用(Kubas interactions))而形成固态氢化物,如氢化物MHx(例如M=钛、钒、铬、锰、铁、钴、镍或铜),其中x是约4到约13(如约4.5到约13或约4.6到约13),例如MH4、MH5、MH6、MH7、MH8、MH9、MH10、MH11、MH12或MH13(例如MH5、MH6、MH7、MH8、MH9、MH10、MH11、MH12或MH13)且可逆地释放氢,从而充当储氢材料。
本文所述的金属氢化物作为块状固体在室温下具有稳定性(即,展现低自燃性且减小的空气敏感性),这是实际储氢的重要特点。
在一个方面中,本发明涉及一种制备金属氢化物(例如适用于储氢应用的金属氢化物)的方法。在一个实施例中,所述方法包含:
(i)将烷基或芳基过渡金属化合物(或其组合)在溶剂(例如有机溶剂)中在氢不存在下加热以形成沉淀物;
(ii)任选地分离沉淀物;
(iii)使沉淀物发生氢化;以及
(iv)任选地分离氢化沉淀物。
在一个实施例中,烷基或芳基过渡金属化合物具有式M1R、M1R2、M1R3或M1R4(或其组合),其中:
M1是过渡金属;以及
每个R基团独立地选自烷基、硅烷化烷基、烯基、芳基烷基、杂芳基以及芳基。在一个优选实施例中,R是硅烷化烷基或芳基。
在一个实施例中,R不含β-氢取代基(例如不含β-氢取代基的有机烷基,如均三甲苯基、新戊基、三甲基硅烷基甲基或苯甲基)。初始烷基或芳基过渡金属化合物可以是单体、二聚物、三聚物、四聚物或聚合物。
在一个实施例中,M1选自钛、钒、铬、锰、铁、钴、镍和铜,以及其组合。在另一个实施例中,M1选自钛、钒、铬、锰、铁、钴和镍,以及其组合。在又另一个实施例中,M1选自钒、锰和铬,以及其组合。
在一个实施例中,步骤(i)的产物含有大于约10重量%(如大于约20重量%、大于约30重量%、大于约40重量%或大于约50重量%)的残余烃。在另一个实施例中,步骤(i)的产物含有小于约50重量%(如小于约40重量%、小于约30重量%、小于约20重量%或小于约10重量%)的残余烃。
在一个实施例中,步骤(i)是在约5℃到约250℃(如约50℃到约200℃、约75℃到约150℃、约80℃到约120℃、约90℃到约110℃或约95℃到约105℃)的温度下进行。在一个实施例中,步骤(i)是在约100℃下进行。
在一个实施例中,步骤(i)进行的时间段在约12小时与约72小时之间,例如在约24小时与约60小时之间,如约24小时或约48小时。
在一个实施例中,步骤(i)是在约100℃的温度下进行约48小时的时段。
在一个实施例中,步骤(i)是在脂肪族溶剂(如烃溶剂,例如戊烷、己烷、环己烷、庚烷、辛烷以及其组合)中进行。在一个实施例中,步骤(i)是在石油醚中进行。在一个实施例中,步骤(i)是在芳香族(例如甲苯)中进行。步骤(i)中的溶剂优选无水溶剂。在一个实施例中,步骤(i)是溶解,随后形成所期望的沉淀物。
在一个实施例中,步骤(ii)包含过滤步骤(i)的产物。在另一个实施例中,步骤(ii)包含过滤步骤(i)的产物,随后干燥所得固体(例如在真空下,任选地在约50℃与200℃之间(如约100℃与150℃之间,例如约100℃)的温度下干燥约1小时与约10小时之间(如约2小时与6小时之间,例如约4小时)的时间段)。在一个实施例中,步骤(ii)包含过滤步骤(i)的产物,随后使所得固体在约100℃的温度下真空干燥约四小时。
在一个实施例中,步骤(iii)中的氢化是在约1巴与约200巴之间(如约25巴与约150巴之间,约50巴与约125巴之间,约50巴与约100巴之间,或约60巴到约80巴)的氢压力进行。在其它实施例中,步骤(iii)中的氢化是在约1巴、约5巴、约10巴、约15巴、约20巴、约25巴、约30巴、约40巴、约50巴、约60巴、约70巴、约80巴、约90巴或约100巴的氢压力下进行。在一个实施例中,步骤(iii)中的氢化是在70巴的氢压力下进行。
在一个实施例中,步骤(iii)是在约10℃到约200℃(如约10℃到约100℃、约15℃到约50℃、约20℃到约40℃、约20℃到约30℃)的温度下进行。在一个实施例中,步骤(iii)是在约25℃下进行。在一个实施例中,步骤(iii)是在室温下进行。在一个实施例中,步骤(iii)是不加热或冷却的情况下进行。
在一个实施例中,步骤(iii)进行的时间段在约12小时与约72小时之间,例如在约24小时与约60小时之间,如约48小时。在另一个实施例中,步骤(iii)进行的时间段在约1天与约7天之间,例如约2天、约3天、约4天、约5天、约6天或约7天。
在一个实施例中,步骤(iii)是在约25℃的温度和约70巴的氢压力下进行约48小时。
在一个实施例中,步骤(iii)是在溶剂不存在下进行。在另一个实施例中,步骤(iii)是在脂肪族溶剂(如烃溶剂,例如石油醚、戊烷、环己烷、己烷、庚烷、辛烷,以及其组合)中进行。在一个实施例中,步骤(iii)是在芳香族溶剂(例如甲苯)中进行。步骤(iii)中的溶剂优选无水溶剂。
在一个实施例中,方法包含步骤(ii)(即,步骤(ii)并非任选的且形成所述方法的一部分)。在另一个实施例中,所述方法包含步骤(iv)(即,步骤(iv)并非任选的且形成所述方法的一部分)。在一个优选实施例中,所述方法包含步骤(i)-(iv)(即,步骤(ii)和(iv)并非任选的且形成所述方法的一部分)。
在另一个实施例中,所述方法进一步包含(v)使步骤(iii)(或步骤(iv)(若执行))的产物进行一个或多个(如约5或更多个、约10或更多个、约20或更多个、约30或更多个、约40或更多个,或约50或更多个)氢吸附-解吸附循环。
在步骤(v)的一个实施例中,氢吸附-解吸附循环可以在约1巴与约250巴之间、约1巴与约200巴之间、约50巴与约170巴之间、约100巴与约150巴之间或约120巴与约150巴之间的氢压力下进行。在其它实施例中,步骤(v)中的氢化是在约1巴、约5巴、约10巴、约15巴、约20巴、约25巴、约30巴、约40巴、约50巴、约60巴、约70巴、约80巴、约90巴、约100巴、约125巴或约150巴的氢压力下进行。
在另一个方面中,本发明涉及一种制备如下文所述的式I或II的金属氢化物的方法,其中所述方法包含上述任一个实施例中所述的步骤。
在另一个方面中,本发明涉及通过根据本文所述的任一个实施例的方法所制备的金属氢化物(例如适用于储氢应用的金属氢化物,如本文所述的任一种金属氢化物)。在另一个方面中,本发明涉及通过根据本文所述的任一个实施例的方法所制备的金属氢化物(例如适用于储氢应用的金属氢化物,如本文所述的任一种金属氢化物),其中金属氢化物能够吸收至少4.0%(例如约4.0%到约12.0%、约5.0%到12.0%,或更高)(按其中未储存氢的金属氢化物的100%总重量计)的量的氢(H2)。
在另一个方面中,本发明涉及式(I)的金属氢化物:
M1(M2)zHxRyLn (I)
其中
M1是选自钛、钒、铬、铁、钴、镍、铜以及其任选混合物的第一金属;
M2是一种或多种其它金属,其具有总含量z(例如一种或多种掺杂金属,比如(例如)锌、镓、锆、铌、钼、锝、钌、铑、钯、银、镉、铪、钽、钨、铼、锇、铱、铂、金以及汞);
R若存在,则是有机基团(例如不含β-氢取代基的有机基团);
L是路易斯碱(例如有机溶剂(如醚溶剂,例如Et2O、二噁烷、THF)、水、H2S、胺、膦、硫化物、烯烃(例如1-己烯)以及其组合);
n是0到约1(例如0到约0.8、0到约0.6、0到约0.5、0到约0.4、0到约0.2、0到约0.1、0到约0.05或0到约0.01);
y是0到约0.5,以及
z是0到约1(例如0到约0.9、0到约0.8、0到约0.7、0到约0.6、0到约0.5、0到约0.4、0到约0.3、0到约0.2、0到约0.1或0到约0.05);
其中
当M1是Ti或V时,x是约4.6到约13;
当M1是Cr时,x是约4.6到约12;
当M1是Fe时,x是约4.6到约10;
当M1是Ni或Co时,x是约4.6到约8;
当M1是Cu时,x是约4.6到约6。
在一个实施例中,M1选自钛、钒、铬、铁,以及其混合物。在一个实施例中,M1选自钛、钒、铬,以及其混合物。在一个实施例中,M1选自钒、铬,以及其混合物。
在另一个实施例中,x是约8.6到约13、约8.6到约12、约8.6到约11,或约8.6到约10。
在一个实施例中,M1是Ti且x大于7(例如约7.5到约13、约8到约13、约9到约13、约10到约13、约11到约13,或约12到约13)。举例来说,M1是Ti且x是约7.5到约8.5、约8.5到约9.5、约9.5到约10.5、约10.5到约11.5、约11.5到约12.5,或约12.5到约13。
在一个实施例中,M1是V且x大于7(例如约7.5到约13、约8到约13、约9到约13、约10到约13、约11到约13或约12到约13)。举例来说,M1是V且x是约7.5到约8.5、约8.5到约9.5、约9.5到约10.5、约10.5到约11.5、约11.5到约12.5,或约12.5到约13。
在一个实施例中,M1是Cr且x大于6(例如约6.5到约12、约7到约12、约8到约12、约9到约12、约10到约12,或约11到约12,a)。举例来说,M1是Cr且x是约6.5到约7.5、约7.5到约8.5、约8.5到约9.5、约9.5到约10.5、约10.5到约11.5,或约11.5到约12。
在一个实施例中,M1是Fe且x大于6(例如约6.5到约10、约7到约10、约8到约10,或约9到约10)。举例来说,M1是Fe且x是约6.5到约7.5、约7.5到约8.5、约8.5到约9.5,或约9.5到约10。
在一个实施例中,M1是Co且x大于6(例如约6.5到约8或约7到约8)。举例来说,M1是Co且x是约6.5到约7.5或约7.5到约8。
在一个实施例中,M1是Ni且x大于6(例如约6.5到约8或约7到约8)。举例来说,M1是Ni且x是约6.5到约7.5或约7.5到约8。
在另一个方面中,本发明涉及式(II)的金属氢化物:
M1(M2)zHxRyLn(H2)a (II)
其中
M1是选自钛、钒、铬、铁、钴、镍、铜以及其混合物的第一金属;
M2是一种或多种其它金属(例如除钛、钒、铬、铁、钴、镍以及铜以外的金属),其具有总含量z(例如一种或多种掺杂金属,比如(例如)锆、镓、锌、铌、钼、锝、钌、铑、钯、银、镉、铪、钽、钨、铼、锇、铱、铂、金以及汞);
R若存在,则是有机基团(例如不含β-氢取代基的有机基团);
L是路易斯碱(例如有机溶剂(如醚溶剂,例如Et2O、二噁烷以及THF)、水、H2S、胺、膦、硫化物、烯烃(例如1-己烯),以及其组合);
n是0到约1(例如0到约0.8、0到约0.6、0到约0.5、0到约0.4、0到约0.2、0到约0.1、0到约0.05或0到约0.01);
x是约0.5到约4.5(例如约1.8到约4.2或约2到约4);
a大于1(如大于2;如约3到约5,例如约3、约4或约5);
y是0到约0.5,以及
z是0到约1(例如0到约0.9、0到约0.8、0到约0.7、0到约0.6、0到约0.5、0到约0.4、0到约0.3、0到约0.2、0到约0.1或0到约0.05);
在一个实施例中,a是约2到约5(例如约2、约3、约4或约5)。在一个实施例中,a是约3到约5(例如约3、约4或约5)。在一个实施例中,a是约3。在一个实施例中,a是约4。在一个实施例中,a是约5。
在一个实施例中,M1是Ti,x是约3且a是约3到约5(例如约3、约4或约5)。
在一个实施例中,M1是V,x是约3且a是约3到约5(例如约3、约4或约5)。
在一个实施例中,M1是Cr,x是约2且a是约3到约5(例如约3、约4或约5)。
在一个实施例中,M1是Fe,x是约2且a是约3到约5(例如约3、约4或约5)。
在一个实施例中,M1是Co,x是约2且a是约3。
在一个实施例中,M1是Ni,x是约2且a是约3。
在另一个方面中,本发明涉及式(III)的金属氢化物:
M1(M2)zHxRyLn (III)
其中
M1是选自钛、钒、铬、铁、钴、镍、铜以及其任选混合物的第一金属;
M2是一种或多种其它金属,其具有总含量z(例如一种或多种掺杂金属,比如(例如)锌、镓、锆、铌、钼、锝、钌、铑、钯、银、镉、铪、钽、钨、铼、锇、铱、铂、金以及汞);
R若存在,则是有机基团(例如不含β-氢取代基的有机基团);
L是路易斯碱(例如有机溶剂(如醚溶剂,例如Et2O、二噁烷、THF)、水、H2S、胺、膦、硫化物、烯烃(例如1-己烯)以及其组合);
n是0到约1(例如0到约0.8、0到约0.6、0到约0.5、0到约0.4、0到约0.2、0到约0.1、0到约0.05或0到约0.01);
y是0到约0.5;
z介于大于0.2与约1之间(例如约0.25到约1、约0.25到约0.9、约0.25到约0.8、约0.25到约0.7、约0.25到约0.6、约0.25到约0.5、约0.25到约0.4、约0.25到约0.3);以及
x是约0.5到约4.5(例如约2.5到约4.5、约0.5到约3.6或约0.5到约3.3)。
在式(III)的金属氢化物的一个实施例中,以下中的一或多种(如2、3或4种)适用:(i)当M1是钒时,x是至少2.4;(ii)当M1是铜时,x是至少1.0;(iii)当M1是钛时,x是至少2.4和/或(iv)当M1是镍时,x是至少1.6。
在本文所述的任一种金属氢化物的一个实施例中,y小于约0.4,如小于约0.3、小于约0.2、小于约0.1或小于约0.05。在一个实施例中,y是0到约0.4,如0到约0.3、0到约0.25、0到约0.2、0到约0.1,或0到约0.05。
在本文所述的任一种金属氢化物的一个实施例中,R若存在,则独立地选自烷基、硅烷化烷基、烯基、芳基烷基、杂芳基以及芳基。在一个优选实施例中,R若存在,则是硅烷化烷基或芳基。
在本文所述的任一种金属氢化物的一个实施例中,金属氢化物的氢化和/或脱氢在热力学上是中性的,如针对块状样品平均化时。举例来说,与氢吸附过程和/或氢解吸附过程相关的净焓变化(如针对块状样品平均化)接近于0kJ mol-1H2。
举例来说,在一个实施例中,本文所述的任一种金属氢化物以约0到约±3kJ mol-1H2(如约0到约±2.5kJ mol-1H2、约0到约±2kJ mol-1H2、约0到约±1.5kJ mol-1H2、约0到约±1kJ mol-1H2、约0到约±0.5kJ mol-1H2或约0到约±0.25kJ mol-1H2)的绝对值吸附氢和/或解吸氢。
在另一个实施例中,本文所述的任一种金属氢化物以约±0.5到约±3kJ mol-1H2(如约±0.5到约±2.5kJ mol-1H2、约±0.5到约±2kJ mol-1H2、约±0.5到约±1.5kJ mol-1H2、约±0.5到约±1kJ mol-1H2或约±0.5到约±0.75kJ mol-1H2)的绝对值吸附和/或解吸氢。
在另一个实施例中,本文所述的任一种金属氢化物以约±1到约±3kJ mol-1H2(如约±1到约±2.5kJ mol-1H2、约±1到约±2kJ mol-1H2、约±1到约±1.5kJ mol-1H2或约±1到约±1.25kJ mol-1H2)的绝对值吸附和/或解吸氢。
在另一个实施例中,本文所述的任一种金属氢化物以约±1.5到约±3kJ mol-1H2(如约±1.5到约±2.5kJ mol-1H2、约±1.5到约±2kJ mol-1H2或约±1.5到约±1.75kJ mol-1H2)的绝对值吸附和/或解吸氢。
在另一个实施例中,本文所述的任一种金属氢化物以小于约±4kJ mol-1H2(如小于约±3.75kJ mol-1H2、小于约±3.5kJ mol-1H2、小于约±3.25kJ mol-1H2、小于约±3kJ mol-1H2、小于约±2.75kJ mol-1H2、小于约±2.5kJ mol-1H2、小于约±2.25kJ mol-1H2、小于约±2kJ mol-1H2、小于约±1.75kJ mol-1H2、小于约±1.5kJ mol-1H2、小于约±1.25kJ mol-1H2、小于约±1kJ mol-1H2、小于约±0.75kJ mol-1H2、小于约±0.5kJ mol-1H2、小于约±0.25kJ mol-1H2或小于约±0.1kJ mol-1H2)的绝对值吸附氢和/或解吸氢。
在另一个实施例中,本文所述的任一种金属氢化物以约±3kJ mol-1H2(如约±2.9kJ mol-1H2、约±2.8kJ mol-1H2、约±2.7kJ mol-1H2、约±2.6kJ mol-1H2、约±2.5kJ mol-1H2、约±2.4kJ mol-1H2、约±2.3kJ mol-1H2、约2.2kJ mol-1H2、约±2.1kJ mol-1H2、约±2kJ mol-1H2、约±1.9kJ mol-1H2、约±1.8kJ mol-1H2、约±1.7kJ mol-1H2、约±1.6kJ mol-1H2、约±1.5kJ mol-1H2、约±1.4kJ mol-1H2、约±1.3kJ mol-1H2、约±1.2kJ mol-1H2、约±1.1kJ mol-1H2、约±1kJ mol-1H2、约±0.9kJ mol-1H2、约±0.8kJ mol-1H2、约±0.7kJ mol-1H2、约±0.6kJ mol-1H2、约±0.5kJ mol-1H2、约±0.4kJ mol-1H2、约±0.3kJ mol-1H2、约±0.2kJ mol-1H2或约±0.1kJ mol-1H2)的绝对值吸附氢和/或解吸氢。
在本文所述的任一种金属氢化物的一个实施例中,金属氢化物呈块体相。在本文所述的任一种金属氢化物的一个实施例中,金属氢化物是聚合物,例如呈块体相的聚合物。
在一个实施例中,本文所述的任一种金属氢化物具有介孔性(例如具有约0.5nm与约50nm之间或约2nm与约50nm之间的孔径)。在另一个实施例中,本文所述的任一种金属氢化物具有微孔性(例如具有小于约2nm的孔径,如小于约1nm)。在一个实施例中,任一种所述金属氢化物具有约2nm的孔径。
在一个实施例中,本文所述的任一种金属氢化物具有约5%与约80%之间的孔隙率,如约5%与约70%之间、约5%与约60%之间、约5%与约50%之间、约5%与约40%之间、约5%与约30%之间或约5%与约20%之间。
在一个实施例中,本文所述的任一种金属氢化物是无定形的或实质上无定形的(例如氢化物结构的原子位置具有极小长程有序(例如纳米观有序)或无长程有序)。在一个实施例中,本文所述的任一种金属氢化物含有小于约20%结晶度,如小于约10%、小于约5%、小于约2.5%、小于约1%、小于约0.5%结晶度,或小于约0.1%结晶度,如通过例如使用Cu Kα辐射(40kV,40mA)源的X射线衍射所测量。
在另一个实施例中,本文所述的任一种金属氢化物(例如式(I)、(II)或(III)的金属氢化物)展现以下特性中的一种或多种(如两种或三种):(i)R当存在于金属氢化物中时通过R基团中的碳原子(如单个碳原子)结合到金属中心;(ii)金属氢化物作为块状固体在室温下具有稳定性以及(iii)金属氢化物能够可逆地吸收且释放氢。
在一个实施例中,本发明涉及一种组合物,其包含一种或多种式(I)金属氢化物:
M1(M2)zHxRyLn (I)
其中
每个M1独立地是选自钛、钒、铬、铁、钴、镍、铜以及其任选混合物的第一金属;
每个M2独立地是一种或多种其它金属,其具有总含量z(例如一种或多种掺杂金属,比如(例如)锌、镓、锆、铌、钼、锝、钌、铑、钯、银、镉、铪、钽、钨、铼、锇、铱、铂、金以及汞);
每个R若存在则独立地是有机基团(例如不含β-氢取代基的有机基团);
每个L独立地是路易斯碱(例如有机溶剂(如醚溶剂,例如Et2O、二噁烷以及THF)、水、H2S、胺、膦、硫化物、烯烃(例如1-己烯)以及其组合);
每个n独立地是0到约1(例如0到约0.8、0到约0.6、0到约0.5、0到约0.4、0到约0.2、0到约0.1、0到约0.05或0到约0.01);
每个y独立地是0到约0.5;以及
每个z独立地是0到约1(例如0到约0.9、0到约0.8、0到约0.7、0到约0.6、0到约0.5、0到约0.4、0到约0.3、0到约0.2、0到约0.1或0到约0.05);
其中
当M1是Ti或V时,x是约4.6到约13;
当M1是Cr时,x是约4.6到约12;
当M1是Fe时,x是约4.6到约10;
当M1是Ni或Co时,x是约4.6到约8;
当M1是Cu时,x是约4.6到约6。
在另一个实施例中,本发明涉及一种组合物,其包含一种或多种式(II)金属氢化物:
M1(M2)zHxRyLn(H2)a (II)
其中
每个M1独立地是选自钛、钒、铬、铁、钴、镍、铜以及其混合物的第一金属;
每个M2独立地是一种或多种其它金属(例如除钛、钒、铬、铁、钴、镍以及铜以外的金属),其具有总含量z(例如一种或多种掺杂金属,比如(例如)锆、镓、锌、铌、钼、锝、钌、铑、钯、银、镉、铪、钽、钨、铼、锇、铱、铂、金以及汞);
每个R若存在则独立地是有机基团(例如不含β-氢取代基的有机基团);
每个L独立地是路易斯碱(例如有机溶剂(如醚溶剂,例如Et2O、二噁烷以及THF)、水、H2S、胺、膦、硫化物、烯烃(例如1-己烯)以及其组合);
每个n独立地是0到约1(例如0到约0.8、0到约0.6、0到约0.5、0到约0.4、0到约0.2、0到约0.1、0到约0.05或0到约0.01);
每个x独立地是约0.5到约4.5(例如约1.8到约4.2或约2到约4);
每个a独立地大于1(如大于2;如约3到约5,例如约3、约4或约5);
每个y独立地是0到约0.5,以及
每个z独立地是,z是0到约1(例如0到约0.9、0到约0.8、0到约0.7、0到约0.6、0到约0.5、0到约0.4、0到约0.3、0到约0.2、0到约0.1或0到约0.05)。
在另一个实施例中,本发明涉及一种组合物,其包含一种或多种式(I)金属氢化物以及一种或多种式(II)金属氢化物:
M1(M2)zHxRyLn (I)M1(M2)zHxRyLn(H2)a (II)
其中
每个M1独立地是选自钛、钒、铬、铁、钴、镍、铜以及其混合物的第一金属;
每个M2独立地是一种或多种其它金属(例如除钛、钒、铬、铁、钴、镍以及铜以外的金属),其具有总含量z(例如一种或多种掺杂金属,比如(例如)锆、镓、锌、铌、钼、锝、钌、铑、钯、银、镉、铪、钽、钨、铼、锇、铱、铂、金以及汞);
每个R若存在则独立地是有机基团(例如不含β-氢取代基的有机基团);
每个L独立地是路易斯碱(例如有机溶剂(如醚溶剂,例如Et2O、二噁烷以及THF)、水、H2S、胺、膦、硫化物、烯烃(例如1-己烯)以及其组合);
每个n独立地是0到约1(例如0到约0.8、0到约0.6、0到约0.5、0到约0.4、0到约0.2、0到约0.1、0到约0.05或0到约0.01);
每个y独立地是0到约0.5;以及
每个z独立地是z是0到约1(例如0到约0.9、0到约0.8、0到约0.7、0到约0.6、0到约0.5、0到约0.4、0到约0.3、0到约0.2、0到约0.1或0到约0.05),其中
对于式(I)的金属氢化物来说:
当M1是Ti或V时,x是约4.6到约13;
当M1是Cr时,x是约4.6到约12;
当M1是Fe时,x是约4.6到约10;
当M1是Ni或Co时,x是约4.6到约8;以及
当M1是Cu时,x是约4.6到约6;
且对于式(II)的金属氢化物来说:
每个x独立地是约0.5到约4.5(例如约1.8到约4.2或约2到约4);以及
每个a独立地大于1(如大于2;如约3到约5,例如约3、约4或约5)。
在另一个实施例中,本发明涉及一种储氢方法,所述方法包含:
提供根据本文所述的任一种实施例的金属氢化物(例如一种或多种式(I)或(II)金属氢化物,或其任何混合物);
向金属氢化物中添加氢;以及
允许氢与金属氢化物配位(例如被金属氢化物吸附)。
在另一个实施例中,本发明涉及一种用储存系统储氢的方法,所述方法包含:
提供含有根据本文所述的任一种实施例的金属氢化物(例如一种或多种式(I)或(II)金属氢化物,或其任何混合物)的系统;
向储存系统的金属氢化物中添加氢;以及
允许氢与储存系统中的金属氢化物配位(例如被金属氢化物吸附)。
在一个实施例中,将氢化物任选地与粘合剂和/或润滑剂(例如无定形碳、石蜡、矿物油,或聚合物,如纤维素或聚丙烯)或其它材料(例如促进造粒过程的无机化合物,如TiO2;金属或金属合金,如Ni)一起压制成团粒形式。在此阶段可并入粘合剂、润滑剂和/或其它材料以使供应到最终形式的材料中的氢中的污染物所诱导的中毒、水解或其它潜在有害反应的影响最小化。还可以添加其它添加剂(例如多孔碳、金属有机架构(MOF)以及共价有机架构(COF))以促进本文所述金属氢化物吸附氢以及解吸附氢的速率。在一个实施例中,氢化物是沉积在蜂窝结构化载体的大孔中。
储存系统(例如储槽)槽可以在储存系统的壁中包含一个或多个开口。流体(如氢气)可以通过一个或多个开口通入以及离开储槽。系统可以进一步包含一个或多个控制流体通过一个或多个开口的阀门。通过打开所述一个或多个阀门且允许流体通过一个或多个开口离开储槽,可以使用一个或多个阀门释放储槽内的压力。系统还可以进一步包含用于添加氢到储存系统中的压缩机(例如I气体压缩机)。
在其它实施例中,储氢方法进一步包含从金属氢化物(例如储存系统中的金属氢化物)中释放氢。在一个实施例中,通过降低储存系统中的氢压力来使氢从金属氢化物中释放。在一个实施例中,通过改变(例如提高)储存系统的温度来使氢从金属氢化物中释放。
本发明的又一实施例涉及一种储氢系统,其包含储存系统以及储存系统内的金属氢化物,其中本文所述的任一种实施例包含金属氢化物(例如式(I)以及(II)的金属氢化物)。
本发明的金属氢化物可以适用于其它应用,例如(但不限于)甲烷吸附、压缩天然气储存、推进剂、蓄电池技术、燃料电池、吸附剂、烯烃聚合催化剂以及传感器。本发明的金属氢化物也可以适用于其它应用,如(但不限于)推进式电动和/或混合动力车辆,以及储存电力,同时连接到电网。在一个实施例中,本发明涉及一种配合燃料电池产生能量的储存系统(其可以是任何规模的且可以是固定的或移动的),所述储存系统包含根据本文所述的任何实施例的金属氢化物于储存系统内。
推进剂是用于移动或推进目标(如喷气式飞机或火箭)的材料。推进剂可以包含燃料以及氧化剂。燃料可以是例如汽油、喷气式飞机燃料或火箭燃料。当本发明的金属氢化物用于推进剂时,推进剂进一步包含氢。氢可以与本发明的金属氢化物中存在的金属中心配位。在一个实施例中,氢呈液体形式。在一个优选实施例中,推进剂进一步包含氧化剂,例如液氧。在一个实施例中,推进剂用于推进喷气式飞机或火箭。在另一个实施例中,其配合氧化剂用于火焰产生装置(比如(例如)焊枪)中。
蓄电池包含一个或多个电化学电池,其使所储存的化学能转化成电能。本发明的金属氢化物可以用于配位以及储存蓄电池中的化合物。在一个优选实施例中,所储存的化合物是氢。在一个实施例中,蓄电池使氢中储存的能量转变成电能。在一个实施例中,本发明的金属氢化物配合燃料电池用于产生电。
吸附剂是一种用于吸收液体或气体的材料。本发明的金属氢化物可以作为吸附剂用于吸收液体或气体。举例来说,本发明的金属氢化物可以用于吸收氢。在一个实施例中,氢呈液体形式。在另一个实施例中,氢呈气体形式。
另一个实施例是使烯烃聚合的包含本发明金属氢化物的催化剂系统。催化剂系统可以进一步包含载体。
又另一个实施例是一种包含使烯烃(例如乙烯、丙烯)聚合或共聚合的方法,所述聚合或共聚合是在本发明的催化剂系统存在下进行。
使用传感器检测物质或测量物理量。传感器产生所述物质已检测到的信号或产生代表物理量的测量结果的信号。信号可以由观察者或由仪器读取。
本发明的金属氢化物可以用于传感器中。举例来说,本发明的金属氢化物可以用于检测氢,例如系统中的氢。在一个实施例中,本发明的金属氢化物度量存在于系统中的氢的量。在一个实施例中,氢呈液体形式。在另一个实施例中,氢呈气体形式。
本发明的金属氢化物可以用于推进电动和/或混合动力车辆或用于储存电力,同时连接到电网。
在另一个方面中,本发明涉及包含根据本文所述任何实施例的金属氢化物的蓄电池或燃料电池。
在另一个方面中,本发明涉及使用燃料电池产生电或使用氧化剂产生热的储存系统,包含储存系统以及根据本文所述任何实施例的金属氢化物。
在另一个方面中,本发明涉及一种用于选自氢、甲烷以及压缩天然气的气体的储存系统,包含储存系统以及根据本文所述任何实施例的金属氢化物。
在另一个方面中,本发明涉及一种使用燃料电池产生电或使用氧化剂产生热的储存系统,包含储存系统以及储存系统内的根据本文所述任何实施例的金属氢化物。
附图说明
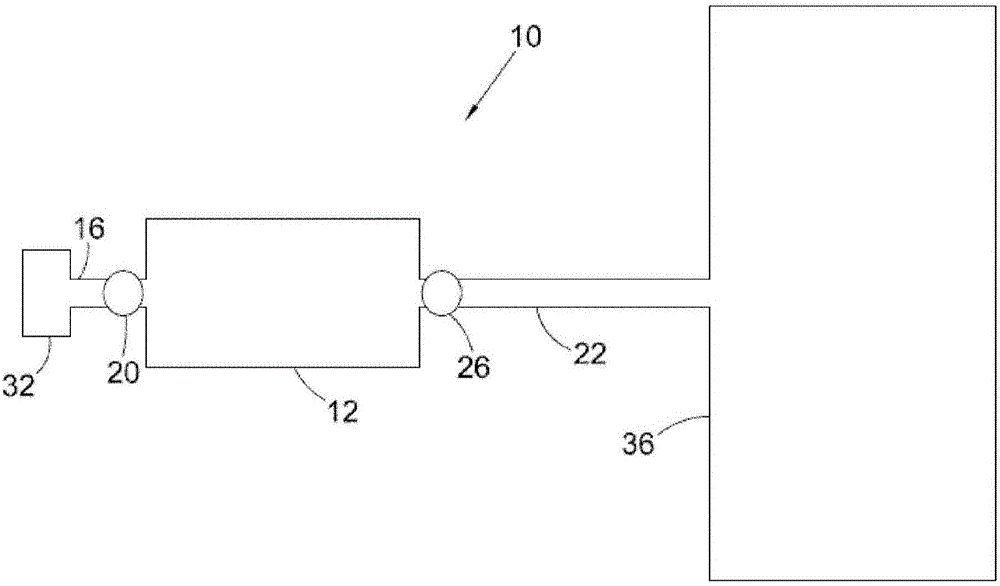
图1描绘适用于本发明的储存系统的一个实施例。
图2描绘附接到氢燃料电池上的储存系统的一个实施例。
图3描绘氢化铬样品Cr-100的IR光谱。
图4描绘氢化铬样品Cr-150C-H2的IR光谱。
图5描绘氢化铬样品Cr-25C-H2的IR光谱。
图6描绘氢化铬样品Cr-100的氮吸附(下部迹线)-解吸附(上部迹线)等温线。
图7描绘氢化铬样品Cr-25C-H2的氮吸附(下部迹线)-解吸附(上部迹线)等温线。
图8描绘氢化铬样品Cr-100的X射线粉末衍射(XRPD)图。
图9描绘氢化铬样品Cr-150C-H2的X射线粉末衍射(XRPD)图。
图10描绘氢化铬样品Cr-25C-H2的X射线粉末衍射(XRPD)图。
图10A和10B描绘氢化铬样品Cr-25C-H2的高分辨率扫描透射电子显微(HTSTEM)影像。
图11描绘氢化铬样品Cr-100的差热分析(DTA)(底部迹线)以及热解重量分析(TGA)(顶部迹线)光谱。
图12描绘氢化铬样品Cr-150C-H2的差热分析(DTA)(底部迹线)以及热解重量分析(TGA)(顶部迹线)光谱。
图13描绘氢化铬样品Cr-25C-H2的差热分析(DTA)(底部迹线)以及热解重量分析(TGA)(顶部迹线)光谱。
图14描绘样品Cr-100的X射线光电子谱(XPS)中的铬2p1/2以及2p3/2区域的峰拟合。
图15描绘样品Cr-100的X射线光电子谱(XPS)中的铬2p1/2以及2p3/2区域的基线校正峰拟合。
图16描绘样品Cr-150C-H2的X射线光电子谱(XPS)中的铬2p1/2以及2p3/2区域的峰拟合。
图17描绘样品Cr-150C-H2的X射线光电子谱(XPS)中的铬2p1/2以及2p3/2区域的基线校正峰拟合。
图18描绘样品Cr-25C-H2的X射线光电子谱(XPS)中的铬2p1/2以及2p3/2区域的峰拟合。
图19描绘样品Cr-25C-H2的X射线光电子谱(XPS)中的铬2p1/2以及2p3/2区域的基线校正峰拟合。
图20描绘氢化铬样品Cr-100、Cr-150C-H2以及Cr-25C-H2在298K下的氢吸附-解吸附等温线。
图21描绘氢化铬样品Cr-25C-H2在0巴与150巴H2之间的10个吸附-解吸附循环的寿命周期氢吸附率(wt.%)。
图22描绘样品Cr-25C-H2在50巴H2(顶部迹线)下以及空样品架在50巴H2(底部迹线)下压缩的拉曼光谱(Raman spectra)。
图23描绘样品Cr-25C-H2在50巴D2(顶部迹线)、50巴H2(中间迹线)下以及在氩气(底部迹线)下的完整拉曼光谱。
图24描绘PCT氢吸附测量时,每次添加H2期间的样品Cr-25C-H2的量热曲线。
图25描绘氢化钒样品V(IV)-100(底部迹线)以及V(IV)-25-H2(顶部迹线)的IR光谱。
图26描绘氢化钒样品V(IV)-100的氮吸附(下部迹线)-解吸附(上部迹线)等温线。
图27描绘氢化钒样品V(IV)-25-H2的氮吸附(下部迹线)-解吸附(上部迹线)等温线。
图28描绘氢化钒样品V(IV)-25-H2的X射线粉末衍射(XRPD)图。
图29描绘氢化钒样品V(IV)-100在298K下的氢吸附(下部迹线)-解吸附(上部迹线)等温线。
图30描绘氢化钒样品V(IV)-25-H2在298K下的氢吸附(下部迹线)-解吸附(上部迹线)等温线。
图31描绘氢化钒样品V(IV)-25-H2在0巴与100巴H2之间的12个吸附-解吸附循环的寿命周期氢吸附率(wt.%)。
图32描绘氢化锰样品Mn(II)-100的IR光谱。
图33描绘氢化锰样品Mn(II)-25C-H2的IR光谱。
图34描绘氢化锰样品Mn(II)-100的氮吸附(下部迹线)-解吸附(上部迹线)等温线。
图35描绘氢化锰样品Mn(II)-25C-H2的氮吸附(下部迹线)-解吸附(上部迹线)等温线。
图36描绘氢化锰样品Mn(II)-100在298K下的氢吸附(下部迹线)-解吸附(上部迹线)等温线。
图37描绘氢化锰样品Mn(II)25C-H2在298K下的氢吸附(下部迹线)-解吸附(上部迹线)等温线。
图38描绘实例4系列1中所述的锰(II)样品的IR光谱。
图39描绘实例4系列1中所述锰(II)样品的IR光谱。
图40描绘实例4系列1中所述的锰(II)样品在暴露于空气2分钟后的IR光谱。
图41描绘实例4系列1中所述的锰(II)样品在暴露于空气5分钟后的IR光谱。
图42描绘实例4系列2中所述的锰(II)样品的IR光谱。
图43描绘实例4系列2中所述的锰(II)样品的IR光谱。
图44描绘实例4系列2中所述的锰(II)样品在暴露于空气5分钟后的IR光谱。
图45描绘实例4系列3中所述的锰(II)样品的IR光谱。
图46描绘实例4系列3中所述的锰(II)样品的IR光谱。
图47描绘实例4系列3中所述的锰(II)样品在暴露于空气2分钟后的IR光谱。
图48描绘实例4系列3中所述的锰(II)样品在暴露于空气5分钟后的IR光谱。
图49描绘氢化锰样品Mn-Cal-150-H2的IR光谱。
图50描绘氢化锰样品Mn-Cal-150-H2在40℃下的氢吸附等温线。
图51描绘PCT氢吸附测量时,每次添加H2期间,氢化锰样品Mn-Cal-150-H2的量热曲线。
图52描绘氢化铬样品Cr(II)-100的IR光谱。
图53描绘氢化铬样品Cr(II)-100的X射线光电子谱(XPS)中的铬2p1/2以及2p3/2区域的峰拟合。
图54描绘氢化铬样品Cr(II)-100的氢吸附-解吸附等温线。
图55描绘氢化铬样品Cr(II)-100的IR光谱。
图56描绘样品V(Mes)-100以及V(Mes)-100H2的IR光谱。
图57描绘样品V(Mes)-100以及V(Mes)-100H2的氢吸附-解吸附等温线。
具体实施方式
定义
除非另外定义,否则本文中所用的所有技术和科学术语通常具有与本发明所属领域的一般技术人员通常所理解相同的含义。
术语“包含”是开放式的且就组合物而言,是指所述成分。如关于本文所述组合物所用的术语“包含”可以替代地涵盖组合物“基本上由所述组分组成”或“由所述组分组成”。
如在此所用的术语“配位”不限于金属中心与氢之间特定类型的相互作用。举例来说,在一个实施例中,金属中心与氢之间的相互作用是库巴斯相互作用(Kubas interaction)。
术语“库巴斯相互作用”是指氢以二氢分子形式、按非解离方式结合到过渡金属中心。在库巴斯相互作用中,金属中心的自由d-电子与氢发生相互作用。具体地说,在金属中心的配位数目较低的情况下,二氢与金属中心共享其σ-键电子,且金属中心通过使其π对称性d-轨道与二氢的空反键σ*空轨道重叠而反向供给电子。由此使得H-H键伸长(不断裂)且位移到H-H共振的较低波数(参见例如《美国化学学会杂志》119,9179-9190,1997)。
不希望受理论所束缚,发明人推测,一个或多个(如2个或更多个,如3、4或5个)H2分子与金属中心通过库巴斯相互作用发生相互作用,形成式MHx的金属氢化物,其中x可以大约是偶数,例如约4、约6、约8、约10或约12。然而,还可以发生双分子和/或自由基过程,产生式MHx的金属氢化物,其中x可以大约是奇数,例如约3、约5、约7、约9、约11或约13。另外,其中变量x是非整数的混合型金属氢化物还可以通过连续(非逐步)吸附来形成。
如本文所用的术语“基本上不含”是指所指定元素或化合物的含量小于约2wt%,如小于约1wt%、小于约0.5wt%、小于约0.1wt%、小于约0.05wt%、小于约0.01wt%、小于约0.005wt%或小于约0.001wt%。
术语“有机基团”是指可以存在于式(I)以及(II)的金属氢化物中的任何含碳基团。举例来说,有机基团可以是用于形成金属氢化物、在合成过程中尚未完全除去的溶剂。有机基团的另一实例可以是在金属氢化物形成期间未完全从金属中心除去的配位体(例如三甲基硅烷基甲基、均三甲苯基、苯甲基或新戊基)。有机基团还可以是添加到金属氢化物中以提高金属氢化物结构的微孔率(例如通过在所述结构内形成桥接甲醇盐配位体)的化合物(例如质子化合物,如甲醇),从而有助于H2移入以及离开金属氢化物。
如本文所用,在一个实施例中,术语“热力学中性”是指针对整个金属氢化物样品平均化时,与氢吸附过程和/或氢解吸附过程相关的净焓变化。举例来说,针对块状样品平均化时,与氢吸附过程和/或氢解吸附过程相关的净焓变化接近于0kJ mol-1H2。典型地,基于显微镜下的氢吸附展现约-5kJ mol-1H2与-70kJmol-1H2之间的焓范围。不希望受理论所束缚,本发明人推测,外部压力打开金属氢化物中的结合位点所必需的能量与放热性M-H键形成过程大约相等且相反,从而产生有效的焓缓冲性以及热力学中性。还在不受理论束缚下,本发明人推测,打开本文所述金属氢化物中的氢结合位点所必需的能量是通过逐渐提高外部氢压力来提供,所述能量的值与涉及氢结合到金属的能量大致相等且相反,从而产生热力学中性,且可以被使无定形结构扭转成有利于氢结合的构形所必需的能量抵消。参见例如Skipper等人,《物理化学杂志C》116,19134,2002。
如本文所用,术语“烷基”是指直链或分支链饱和烃部分。在一个实施例中,烷基是直链饱和烃。除非另外说明,“烷基”或“亚烃基”含有1到24个碳原子。代表性饱和直链烷基包括例如甲基、乙基、正丙基、正丁基、正戊基以及正己基。代表性饱和分支链烷基包括例如异丙基、仲丁基、异丁基、叔丁基、新戊基以及异戊基。在一个优选实施例中,“烷基”不含β氢取代基。
如本文所用,术语“芳基”是指通过金属-碳键结合到金属中心的具有6到24个碳原子的芳香族烃(单环或多环)(例如苯基、萘基)。
如本文所用,术语“芳基烷基”是指烷基-芳基,其中烷基以及芳基如本文所定义(例如苯甲基)。
如本文所用,术语“杂芳基”是指具有5到24个碳原子、另外含有一个或多个N、S或O原子的芳族基团(单环或多环)。
所属领域的一般技术人员容易理解,具有非整数化学计算量的金属氢化物(如MH4.2)是指金属(M)原子与不同量的氢配位(例如1份MnH6平均9份MH4)的材料。另外,具有整数化学计算量的金属与氢化物配位体(例如MHx)的本文中所定义的任何金属氢化物还希望涵盖具有整体混合式化学计算量的MH(x-0.2到x+0.2)(例如对于MH11以及MH12来说,分别是MH10.8-11.2或MH11.8-12.2)的金属氢化物样品。
金属氢化物
在一个实施例中,本文所述的任一种金属氢化物具有小于约5m2/g的BET表面积,如小于约4m2/g,如小于约3m2/g、小于约2m2/g、小于约1.5m2/g或小于约1.0m2/g,如约0.6m2/g。
在另一个实施例中,本文所述的任一种金属氢化物具有约2m2/g或更大的BET表面积,如约5m2/g或更大、约7.5m2/g或更大、约10m2/g或更大、约25m2/g或更大、约50m2/g或更大、约75m2/g或更大、约100m2/g或更大、约150m2/g或更大、约200m2/g或更大、约250m2/g或更大、约275m2/g或更大、约300m2/g或更大、约350m2/g或更大、约400m2/g或更大、约450m2/g或更大或约500m2/g或更大。举例来说,金属氢化物具有约377m2/g或391m2/g的BET表面积。
在其它实施例中,BET表面积是约2m2/g到约1000m2/g,如约10m2/g到约750m2/g、约50m2/g到约500m2/g、约100m2/g到约500m2/g、约250m2/g到约500m2/g、约300m2/g到约500m2/g。在一个实施例中,BET表面积是约300m2/g到约400m2/g。
在一个实施例中,本文所述的金属氢化物呈凝胶形式。在一个实施例中,本文所述的金属氢化物呈固体(例如粉末)形式。在一个实施例中,本文所述的任一种金属氢化物是块状固体,例如在室温下稳定的块状固体。在一个实施例中,本文所述的金属氢化物是聚合物(例如呈块体相的聚合物)。在一个实施例中,本文所述的金属氢化物呈团粒形式。
在一个实施例中,任一种所述金属氢化物具有约2nm的孔径。
在一个实施例中,本文所述的任一种金属氢化物具有约5%与约80%之间的孔隙率,如约5%与约70%之间、约5%与约60%之间、约5%与约50%之间、约5%与约40%之间、约5%与约30%之间或约5%与约20%之间。
在其它实施例中,按其中未储存分子氢的金属氢化物的100%总重量计,本文所述的任一种金属氢化物展现至少约2%、至少约3%、至少约4%、至少约5%、至少约6%、至少约7%、至少约8%、至少约9%、至少约10%、至少约11%、至少约12%、至少约13%或至少约14%的重量氢吸附量,例如量高达约14%,如约2.0%到约14.0%、约8.0%到约12.0%,或约3.5%、约7.0%、约10.5%、约14%)。
在一个实施例中,有机基团R不含β氢取代基(例如R是均三甲苯基、新戊基、苯甲基或三甲基硅烷基甲基)。在一个优选实施例中,R是新戊基或均三甲苯基。
在另一个实施例中,本文所述的任一种金属氢化物不含或实质上不含金属离子(除钛、钒、铬、铁、钴、镍和/或铜以外)。在另一个实施例中,本文所述的任一种金属氢化物不含或基本上不含有机残余物(例如在金属氢化物或其前驱物合成期间所用的有机配位体或溶剂)。在另一个实施例中,本文所述的任一种金属氢化物不含或基本上不含金属离子(除钛、钒、铬、铁、钴、镍和/或铜以外)且不含或基本上不含有机残余物(例如在金属氢化物或其前驱物合成期间所用的有机配位体或溶剂)。
在另一个实施例中,本文所述的任一种金属氢化物可以含有超过一种氧化态的过渡金属(例如M(I)/M(II)、M(I)/M(III)、M(II)/M(IV)、(M(II)/M(III)、M(II)/M(0)),其中M是如本文所述的金属。
在一个实施例中,本文所述的任一种金属氢化物包含大于约25wt.%的MHx(其中x如本文任何实施例中所述),如大于约30wt.%、大于约40wt.%、大于约50wt.%、大于约60wt.%、大于约70wt.%、大于约75wt.%、大于约80wt.%、大于约85wt.%、大于约90wt.%、大于约95wt.%、大于约99wt.%、大于约99.5wt.%的MHx。
在本文所述任一种金属氢化物的一个实施例中,金属氢化物中的M-H(金属-氢)键与M-C(金属-碳)键的比率大于约2∶1,如大于约2.5∶1、大于约5∶1、大于约10∶1、大于约20∶1、大于约25∶1、大于约50∶1、大于约75∶1、大于约100∶1、大于约250∶1。
在本文所述任一种金属氢化物的一个实施例中,金属氢化物能够与H2配位。举例来说,在本文所述任一种金属氢化物的一个实施例中,金属氢化物能够与H2通过库巴斯相互作用来配位。
本文所述的金属氢化物优选具有足以允许H2移入以及离开金属氢化物构架到活性结合位点的微孔率(其可以或可以不通过氮吸附来显示)。在一个实施例中,本发明涉及一种包含本文所述任一种实施例的金属氢化物的金属氢化物储存材料,其中所述材料的微孔率足以允许:(i)H2扩散入以及离开材料以及金属氢化物活性结合位点;(ii)金属通过例如库巴斯相互作用而与H2配位;以及(iii)以约2.0%到约14.0%的量(按其中未储存氢的金属氢化物的100%总重量计)吸附H2。金属氢化物储存材料可以并入如本文所述的储氢系统中。
在又另一个实施例中,本文所述的任一种金属氢化物是结晶体。在一个实施例中,且不受理论束缚,H2可以通过穿梭机制移动通过所述结构,借此其在一侧结合到金属且在另一侧解吸附以进一步渗入结构中,或移动通过结晶体平面之间的薄片。
在一个实施例中,本文所述的金属氢化物是无定形的或实质上无定形的(例如氢化物结构中的原子位置具有极小长程有序(例如纳米观有序)或无长程有序)。在一个实施例中,本文所述的金属氢化物含有小于约20%结晶度,如小于约10%、小于约5%、小于约2.5%、小于约1%、小于约0.5%结晶度,或小于约0.1%结晶度,如通过例如使用Cu Kα辐射(40kV,40mA)源的X射线衍射所测量。具有闭合堆积结构的金属氢化物由于其体积密度较高而是所期望的,只要其允许H2扩散到其内的金属结合位点。在金属氢化物的闭合堆积结构不允许H2扩散到金属结合位点的情况下,金属氢化物优选不具有闭合堆积结构。
在一个实施例中,本文所述的金属氢化物是大于80%无定形的,如大于约85%、大于约90%、大于约95%、大于约99%或大于约99.5%无定形的,如通过例如使用Cu Kα辐射(40kV,40mA)源的X射线衍射所测量。
在另一个实施例中,本文所述的任一种金属氢化物可以含有少量(例如总计至多0.5摩尔)的杂质,所述杂质选自膦(例如三甲基膦)、醚、水、醇、胺、烯烃、硫化物、氮化物以及其组合。膦(例如三甲基膦)、醚、水、醇、胺、烯烃(例如1-己烯)、硫化物或氮化物残余物可以从其用于合成金属氢化物时就存在或可以在合成期间作为副产物形成。在一个实施例中,本发明的任一种金属氢化物可以含有小于约10.0wt%、小于约9.0wt%、小于约9.0wt%、小于约7.5wt%、小于约5.0wt%、小于约4.0wt%、小于约3.0wt%、小于约2.0wt%、小于约1.0wt%、小于约0.75wt%、小于约0.5wt%、小于约0.4wt%、小于约0.3wt%、小于约0.25wt%、小于约0.2wt%、小于约0.1wt%、小于约0.05wt%、小于约0.01wt%、小于约0.005wt%或小于约0.001wt%的膦(例如三甲基膦)、醚(例如Et2O、THF、二噁烷)、水、醇、胺、烯烃(例如1-己烯)、硫化物或氮化物残余物,或其组合。在一个优选实施例中,金属氢化物不含或基本上不含膦(例如三甲基膦)、醚、水、醇、胺、烯烃、硫化物或氮化物残余物,或其组合。另外,在发现杂质的本发明实施例中,本文所述的金属氢化物也可以含有少量(例如总计至多0.5摩尔)的金属氢氧化物(M-OH)以及金属醚(M-O-M),其是反应混合物内所含的残余水使金属烷基物质水解所产生。
在某些实施例中,本发明的任一种金属氢化物含有小于约10.0wt%的锂或镁,或其组合。这些锂以及镁残余物可以从其用于合成金属氢化物时就存在。举例来说,本发明的任一种金属氢化物可以含有小于约9.0wt%、小于约8.0wt%、小于约7.5wt%、小于约5.0wt%、小于约4.0wt%、小于约3.0wt%、小于约2.0wt%、小于约1.0wt%、小于约0.75wt%、小于约0.5wt%、小于约0.25wt%、小于约0.1wt%或小于约0.05wt%、小于约0.01wt%、小于约0.005wt%或小于约0.001wt%的锂或镁或其组合。在另一个实施例中,本发明的任一种金属氢化物含有小于约0.5wt%的锂或镁,或其组合。举例来说,本发明的任一种金属氢化物可以含有小于约0.4wt%、小于约0.3wt%、小于约0.25wt%、小于约0.2wt%、小于约0.1wt%、小于约0.05wt%、小于约0.01wt%、小于约0.005wt%或小于约0.001wt%的锂或镁,或其组合。在一个优选实施例中金属氢化物不含或基本上不含锂或镁,或其组合。
本发明的金属氢化物可以含有卤素。举例来说,金属氢化物可以含有小于约20.0wt%的卤素,如小于约10.0wt%的卤素(如Br-、Cl-或I-)。这些卤素残余物可以从其用于合成金属氢化物时(例如从使用格林纳试剂(Grignard reagent)时)就存在。举例来说,本发明的任一种金属氢化物可以含有小于约9.0wt%、小于约8.0wt%、小于约7.5wt%、小于约5.0wt%、小于约4.0wt%、小于约3.0wt%、小于约2.0wt%、小于约1.0wt%、小于约0.75wt%、小于约0.5wt%、小于约0.25wt%、小于约0.1wt%、小于约0.05wt%、小于约0.01wt%、小于约0.005wt%或小于约0.001wt%的卤素。在一个优选实施例中,金属氢化物不含或基本上不含卤素。
在一个实施例中,本文所述的任一种金属氢化物(例如式(I)以及(II))能够以至少2.0%(例如约2.0%到约8.0%、约8.0%到约12.0%,或更高)的量(按其中未储存氢的金属氢化物的100%总重量计)吸收分子氢(H2)。在另一个实施例中,金属氢化物能够以约2.0%、2.5%、3.0%或3.5%到约4.0%、4.5%、5.0%、5.5%、6.0%、6.5%或7.0%的量吸收分子氢(H2)。在另一个实施例中,金属氢化物能够以约8.0%、8.5%或9.0%到约11.0%、11.5%或12%的量吸收分子氢(H2)。
按其中未储存分子氢的金属氢化物的100%总重量计,本发明的金属氢化物能够以至少约2%、至少约3%、至少约4%、至少约5%、至少约6%、至少约7%、至少约8%、至少约9%、至少约10%、至少约11%、至少约12%、至少约13%或至少约14%的量吸收分子氢(H2),例如高达约14%的量,如约2.0%到约14.0%、约8.0%到约12.0%,或约3.5%、约7.0%、约10.5%、约14%)。
在本文所述的任一种金属氢化物的一个实施例中,R若存在,则选自烷基、硅烷化烷基、烯基、芳基烷基、杂芳基以及芳基。
在本文所述任一种金属氢化物的一个实施例中,R若存在,则选自(三甲基硅烷基)甲基、双(三甲基硅烷基)甲基、苯基、苯甲基、均三甲苯基(或下述式(V)基团,其中R1是任何有机基团)、烯丙基、1,3-二甲基烯丙基、1,3-二乙基烯丙基(或另一个1,3-二取代烯丙基,其中取代基是有机基团)、新戊基、2,2,2-二甲基苯基丙基、苯甲基、在芳族环上被一个或多个基团(例如甲氧基或降冰片烷)在其间位以及对位取代的苯甲基、芳基、在芳族环上被一个或多个基团(例如甲氧基或降冰片烷)在其间位以及对位取代的芳基,以及其组合。在一个实施例中,R是硅烷化烷基,如双(三甲基硅烷基)甲基。
在另一个实施例中,R若存在,则选自(三甲基硅烷基)甲基、双(三甲基硅烷基)甲基、均三甲苯基、烯丙基、1,3-二甲基烯丙基、苯甲基、苯基以及其组合。在一个实施例中,R是硅烷化烷基,如双(三甲基硅烷基)甲基。在一个实施例中,R是苯基。
在一个实施例中,本发明涉及一种金属氢化物储存材料,其包含一种或多种通过本文所述的任一种方法制备的金属氢化物。
在另一个实施例中,本发明涉及一种金属氢化物储存材料,其包含一种或多种根据本文所述的任一种实施例的金属氢化物。
金属氢化物储存材料可以并入如本文所述的储氢系统中。
储氢
在另一个实施例中,本发明涉及一种储氢方法,其包含提供根据本文所述任一种实施例的金属氢化物(例如根据本文所述任一种方法制备的金属氢化物和/或一种或多种式(I)或(II)金属氢化物);向金属氢化物中添加氢;以及允许氢与金属氢化物配位。氢的储存可以用储存系统执行。
适用于储氢的储存系统的一个实施例是压力容器。举例来说,压力容器可以在最高200℃的温度(例如约-100到约150℃、约-50到约0℃、约-25到约0℃、约0到约150℃、约0到约50℃、约10到约30℃或约20到约25℃)下容纳本发明的金属氢化物。在一个实施例中,储存系统基本上不含氧。
氢可以添加到储存系统(例如压力容器)中且使用本发明的金属氢化物储存。在一个实施例中,当氢添加到压力容器中储存时,不需要加热。
可以通过本发明金属氢化物储存的氢的量与储存系统中的压力成比例。举例来说,在较高压力下,本发明的金属氢化物可以储存更多的氢。储存系统中的压力可以通过向储存系统中添加氢来提高。不希望受任何特定理论束缚,本发明人推测,当压力增加时,每个金属中心发生库巴斯相互作用的次数可以增加。举例来说,当金属氢化物是金属二氢化物(如MH2)时,一个氢分子与金属配位(例如通过库巴斯相互作用)而产生MH4。两个氢分子与金属配位(例如通过库巴斯相互作用)而产生MH6。三个氢分子与金属配位(例如通过库巴斯相互作用)而产生MH8。四个氢分子与金属配位(例如通过库巴斯相互作用)而产生MH10。五个氢分子与金属配位(例如通过库巴斯相互作用)而产生MH12。然而,如上文所提到,此过程在块体状态下呈现连续性,从而形成含有具有配位氢分子混合物的金属氢化物的块体材料,且因此,产生锰与氢的总体非整数化学计算量。另外,有可能(例如通过自由基和/或双分子过程)形成式MH3、MH5、MH7、MH9以及MH11的分子物质。
在其它实施例中,本文所述的任一种金属氢化物任选地含有一种或多种其它金属(例如除钛、钒、铬、锰、铁、钴、镍以及铜以外的金属)。举例来说,金属氢化物可以含有一种或多种选自钠、钾、铝、铍、硼、钙、锂、镁以及其组合的其它金属。在替代实施例中,金属氢化物可以含有一种或多种其它金属(例如除钛、钒、铬、锰、铁、钴、镍以及铜以外的金属),其中一种或多种其它金属是氢处理后形成氢化物的周期4、5、6、7、8、9、10、11和/或12过渡金属或镧系元素。举例来说,金属氢化物可以含有一种或多种选自锆、铌、钼、锝、钌、铑、钯、银以及其组合的其它金属。在一个实施例中,本文所述的任一种金属氢化物可以任选地含有一种或多种其它周期4、周期5或周期6过渡金属。在另一个实施例中,金属氢化物可以含有一种或多种选自铁、锆、铌、钼、锝、钌、铑、钯、银、镉、铪、钽、钨、铼、锇、铱、铂、金、汞以及其组合的其它金属。一种或多种其它金属的存在量可以是约50wt.%或更小、约40wt.%或更小、约30wt.%或更小、约25wt.%或更小、约20wt%或更小、约10wt%或更小、约5wt%或更小、约1wt%或更小、约0.75wt%或更小、约0.5wt%或更小、约0.25wt%或更小、约0.1wt%或更小、约0.05wt%或更小,或约0.01wt%或更小。在一个实施例中,本文所述的金属氢化物不含其它金属(例如不含除锰以外的金属)。
系统中的氢气压力可以使用将氢抽入系统中的压缩机(如气体压缩机)增加。系统中的氢气压力优选增加到约30atm或更大。举例来说,系统中的氢气压力可以从约30atm增加到约500atm,从约50atm增加到约200atm,或从约75atm增加到约100atm。
系统优选具有最高200℃的温度(或在最高200℃的温度下操作),如约-200℃到150℃(例如约-100℃到150℃)、约-200℃到100℃、约0℃到50℃、约10℃到30℃,或约20℃到25℃。在一个实施例中,系统具有约25℃到约50℃的温度(或在约25℃到约50℃的温度下操作)。系统优选不含氧以防止系统中的金属氧化。在一个实施例中,在本发明系统中储存以及释放氢的方法可以在不加热系统和/或冷却系统的情况下执行。在另一个实施例中,在本发明系统中储存以及释放氢的方法可以通过加热和/或冷却系统来执行。
在另一个实施例中,使氢从储存系统中释放。举例来说,这可以通过降低系统中的氢压力来完成。在一个实施例中,不需要加热便可使氢从金属氢化物中释放。举例来说,可以打开储存系统中的阀门以允许氢气从系统中逸出,从而降低储存系统中的压力。在一个实施例中,约100%的储氢被释放。在其它实施例中,大于约50%、大于约55%、大于约60%、大于约70%、大于约75%、大于约80%、大于约90%、大于约95%、大于约97.5%、大于约99%或大于约99.5%的氢被释放。释放系统中的氢气压力的步骤可以通过允许氢气从系统中逸出来执行,从而降低氢气压力。举例来说,释放氢气压力的步骤可以使系统中的氢气压力降低到100atm或更小(如50atm或更小、30atm或更小,或20atm或更小)。在另一个实施例中,通过提高系统温度来使氢从储存系统中释放。
可以在系统的整个压力梯度的任一点添加或从系统释放氢,而对系统的储存能力无任何不利影响。在某些实施例中,可以添加氢或从系统中释放氢任意次数,而对系统的储存能力无任何不利影响。举例来说,系统可以填充氢以及清空氢至少100次,如至少200次、至少500次、至少1000次或至少1500次,而不会显著降低系统的储存能力。
在一个实施例中,储存系统(例如压力容器)是车辆(如卡车或机动车)的油箱。
图1描绘适用于本发明的储存系统的一个实施例。图2描绘附接到氢燃料电池上的储存系统的一个实施例。系统10包含槽体12,所述槽体是由不透氢气的材料制成,从而防止氢气非所期望地渗离槽体12。举例来说,槽体12是由金属(比如(例如)钢或铝)制成。或者,槽体12是由复合材料(如玻璃纤维与芳纶复合物)制成。在另一个实施例中,槽体12是由碳纤维与内衬制成。内衬可以是聚合物内衬,如热塑性内衬或金属内衬,如钢内衬或铝内衬。
本发明的金属氢化物14存在于槽体12内部。图1中,金属氢化物14呈凝胶形式。金属氢化物14可以部分地填充或填满槽体12。在某些实施例中,金属氢化物可以作为涂层存在于载体上或以团粒形式存在,这取决于槽体中的压降要求。在其它实施例中,金属氢化物可以与增强涂层或团粒的结构完整性以及其它特性的其它化合物(如粘合剂)混合存在。
第一通道16通向槽体12的壁中的第一开口18。第一阀门20控制氢气通过第一开口18的流动。
第二通道22从槽体12的壁中的第二开口24延伸。第二阀门26控制氢气通过第二开口24的流动。
第一阀门20以及第二阀门26可以是控制氢气分别通过第一开口18以及第二开口24流动的任何类型的阀门。举例来说,第一阀门20以及第二阀门26可以是球阀或闸阀。
在一个实施例中,氢如下添加到系统10中。气体压缩机32将氢气抽入第一通道16中。打开第一阀门20以允许氢气流经第一开口18且流入槽体12。
通道管28与第一开口18处于气态连通且延伸到槽体12的内部。通道管28促进氢气分布到金属氢化物14中。在一个实施例中,通道管28是由氢气可渗透的材料制成。由此允许氢气传递通过通道管28的壁且与金属氢化物14接触。通道管还优选由金属氢化物14不可渗透的材料制成,从而防止金属氢化物14进入通道管28的内部。通道管28优选通向槽体12的内部。通道管28的开口优选用防止金属氢化物14进入通道管28内部的过滤器30覆盖。
当压缩机32将氢气抽入槽体12中时,槽体12内部的氢气压力提高。当槽体内部的氢气压力提高时,金属氢化物14能够与更多量的氢配位。优选地,压力提高使得金属氢化物14中每个金属中心发生库巴斯相互作用的次数增加。期望量的氢已经添加到系统中之后,关闭阀门20。
必要时,可以如下从系统10中释放氢。打开第二阀门26,从而使氢气通过第二开口24流出槽体12。当氢气通过第二开口24流出槽体时,槽体12内部的压力降低。当槽体12内部的压力降低时,金属氢化物14释放氢。举例来说,压力降低可以使得金属氢化物14中每个金属中心发生的库巴斯相互作用次数减少。
金属氢化物14释放的氢可以通过第二开口24流出槽体12。如图2所示,氢可以通过第二通道22流到燃料电池36。燃料电池36优选使用氢作为燃料且使用氧作为氧化剂来产生电。典型地,过滤器存在于第二开口24中,以防止下游微粒损失。
在一个替代性实施例中,本发明的储存系统包含具有单个开口的储槽。在此实施例中,氢通过单个开口流入与流出储槽。使用阀门控制氢通过开口的流动。因为H2结合焓对于热力学中性来说是适中的且可以根据压力来控制结合,所以不同于许多之前的储氢系统,储槽在大部分应用中可以不需要外来的热管理系统。
在一个实施例中,系统是便携式系统。因而,系统可以运输到填充站以填充氢。填充氢之后,然后可以将系统运输到使用氢能的地点。此系统的应用包括(但不限于)车辆、飞机、家庭、建筑物以及野餐。
实例
现将借助于以下非限制性实例进一步描述本发明。应用这些实例的揭露内容时,应该清楚地意识到,所述实例仅仅是说明本发明且不应该理解为以任何方式限制本发明的范围,本发明所涵盖的许多变体以及等效物对于所属领域的技术人员在阅读本发明后将变得显而易见。
所有化学物质均购自Sigma-Aldrich且不经进一步提纯即使用。使用标准施兰克技术(Schlenk techniques)且在氩气手套箱中以及在施兰克氮气管线上执行操控。
在Micromeritics ASAP 2020TM上,在77K下收集氮吸附以及解吸附数据。
在使用KBr圆片的Perkin Elmer Spectrum RX1上执行红外光谱测定。分析之前,IR级KBr在120℃下烘干整夜以除去任何残余水。在手套箱中,用烘干的研杵和研钵研磨空白KBr样品且然后在空气中压缩以形成圆片。获取空白KBr圆片的背景。将约5mg样品与200mg IR级的烘干KBr一起粉碎,且压缩而形成圆片。从样品的IR中减去KBr的光谱。
在10.00℃/min(直至650℃)的干燥空气的流动下,用Netzsch的STA449C分析仪执行热解重量分析(TGA)以及差热分析(DTA)。还使用氩气保护其余部分。
通过将少量粉末放置于小毛细管(直径1mm)中来收集X射线粉末衍射(XRPD)图且使用Co Kα辐射、使用具有Vantec 500 2D检测器的Bruker Discover衍射仪获取XRD光谱。使用0.2mm准直仪限制X射线束。
使用Hy-Energy制造的计算机控制气体吸附Sieverts装置获得氢吸附等温线。使用购自Air Liquide的高纯度氢(6级,99.9999%纯度)。将不锈钢隔片连同材料一起添加到样品架中以减少多余的空隙空间。样品的空隙空间是通过使用各5个吸附和解吸附点(总共10个)在298K下执行氦体积校准来计算,其中丢弃外围值且再操作。对200mg标准AX-21样品(0.65wt.%,70巴以及298K)执行过量氢储存的测量,且确保仪器正确发挥作用以及确保等温线的准确度。碳-AX21的重量储氢能力报道值在35巴下是0.3wt%(Bernard等人,《对不同碳的储氢的评估(Assessment of Hydrogen Storage on Different Carbons)》,IEA报告,2001)。这对应于70巴下的0.6wt%,在200mg样品规模下,其在100巴H2下产生±0.07wt%((0.65-0.6)×100/70)的误差。选择此样品规模,以便吸附绝对量等效于本文所述的金属氢化物储氢实验中的绝对量(约1mmol H2)以消除系统误差,因为仪器测量吸附氢总摩尔数且然后将其换算为wt%。
使用Renishaw in Via Raman显微镜,使用488nm激发激光(对样品施加20mW功率)获得拉曼光谱。将样品放入铝盘中且装载到氩气手套箱(MBraun Labstar)内的样品池中,其中水以及氧含量保持低于0.1ppm。使用显微镜物镜将激光束聚焦到样品上,光点直径为约50μm。使用具有3mm厚蓝宝石窗的订制样品池载物台测量压力相关性拉曼光谱(直至50巴)。使用计算机控制的质量流量控制器以及背压调节器维持氢(99.9995%纯度)或氘(100%)压力。通过采用750次扫描以及增加强度来获得光谱。
使用Setaram制造的反应、扫描以及等温线C80量热计收集热量测定数据。使用两个高压池,一个用于样品且一个用作参考且所述池使用Swagelok制造的连接件、通过不锈钢气体管线连接到PCT-Pro。通过测量540mg钯的氢吸附焓来校准仪器装置。钯的PCT氢吸附测量(直至6巴)与170℃下的等温量热测量同时进行。总钯吸附热经测定为31.6kJ mol-1H2,其符合文献值。测量时,将200mg样品放入具有不锈钢隔片的样品池中以减少空隙空间。还将相同隔片放入参考池中。测量之前,利用氦体积校准来测定池以及气体管线的空隙空间,一个在室温下测定且一个使用40℃的C80熔炉中的池。氢吸附测量是使用PCT-Pro装置设置,氢气每次添加到样品中的投料时间设定为60分钟。这样允许在氢吸附测量值移到下一次投料之前达到热平衡。设定C80量热计以在PCT测量期间对样品的热流进行等温测量,其中熔炉温度在40℃保持恒定。在对空白池进行氢吸附测量的同时,还在40℃下对空白池进行量热法测量。由此测定气体当在不同压力下引入池中时升温期间的热流。从样品的总热量中减去空白试验的总热量。为了测定氢吸附焓,然后将此值除以材料所吸附氢的总摩尔数。
高分辨率扫描透射电子显微法(HRSTEM)是利用日立(Hitachi)的HD-2700专用扫描透射型电子显微镜(STEM),使用装备有CEOS Cs校正器且在200kV下操作的冷场发射体执行。将粉末样品在氩气填充的手套箱中干式沉积于Cu网上,所述铜网被碳膜(Quantifoil)覆盖且具有直径1.2微米的周期性孔。在三种不同模式下进行观察:明场(BF)、高角环形暗场(HAADF)以及二次电子(SE)。
实例1:氢化铬(III)样品
合成
四(三甲基硅烷基甲基)铬的制备
向CrCl3(THF)3(7.617g,20.33mmol)于40-60℃石油醚中的搅拌悬浮液中添加(三甲基硅烷基)甲基锂(76.4mmol,76.64mL于戊烷中的1.0M溶液)。浆液颜色紧接着变为暗紫色。在室温下搅拌混合物3小时且接着过滤,且残余物用三份石油醚(每份10mL)洗涤。将暗紫色滤液浓缩且在室温下真空干燥24小时,得到暗紫色结晶固体(4.8g,96%产率)。参见Schulzk等人,《有机金属化合物(Organometallics)》,21,3810,2002。
氢化铬(III)的形成
将四(三甲基硅烷基甲基)铬(IV)(1.2040g,5.22mmol)于50mL石油醚中的暗紫色溶液放入600mL搅拌式氢化容器中。将容器缓慢加热到100℃且搅拌反应物两天。过滤反应混合物,得到黑色沉淀物以及褐色滤液。沉淀物在100℃下真空干燥四小时,得到对空气湿气敏感的黑色固体(Cr-100)。将此材料放入Hy-Energy PCT-Pro Sieverts装置的不锈钢样品架中且样品架在85巴H2压力下加热到150℃维持四小时。然后将材料冷却到100℃且在此温度下抽真空两小时,得到样品Cr-150C-H2。然后将材料放入600mL不锈钢氢化容器中且将容器在25℃加压到70巴H2维持2天。从压力容器中移出之后,将材料在100℃下真空干燥四小时,对空气以及湿气敏感的黑色固体(Cr-25C-H2)。
样品表征
样品Cr-100、Cr-150-H2以及Cr-25C-H2的红外(IR)光谱分别展示于图3、4以及5中。对于样品Cr-100来说,观察到位于2950以及2897cm-1的C-H片段。1250cm-1的片段可以归因于材料中所存在的(三甲基硅烷基)甲基配位体的C-Si片段。每次氢化处理之后,2950cm-1的C-H与1250cm-1的C-Si片段的强度均降低,原因是氢解期间,烃配位体被氢化物替换。
在77K下所记录的样品Cr-100与Cr-25C-H2的氮吸附-解吸附等温线分别展示于图6以及7中。样品Cr-100与Cr-25C-H2均具有类型2等温线。样品Cr-100具有377m2/g的BET表面积。室温氢化之后,样品Cr-25C-H2的BET表面积增加到391m2/g。这可以归因于烃配位体的去除,从而在结构中产生有助于气体扩散通过结构的新多孔路径。在两种样品中,吸附与解吸附等温线之间存在滞后,这意味着材料是多孔材料。0与0.1P/Po之间存在相当陡峭的增加,表明可能存在一些含量的微孔隙,其占所吸附总体积的约20%。0.1与0.8P/Po之间适中增加的斜率归因于介孔隙且0.8与1.0P/Po之间增加的斜率归因于构造上的多孔性。
样品Cr-100、Cr-150C-H2以及Cr-25C-H2的X射线粉末衍射(XRPD)图分别展示于图8、9以及10中。所有三种样品基本上是无定形的。在图9以及10中,在20-30°区域中可以见到少量反射,其对应于玻璃毛细管。
图10A和10B描绘氢化铬样品Cr-25C-H2的高分辨率扫描透射电子显微(HTSTEM)影像。图10A展示粉末形态且图10B展示氢化铬材料的约2nm孔隙结构。未观察到氢化铬样品Cr-25C-H2的结晶区域。
对样品Cr-100、Cr-150C-H2以及Cr-25C-H2进行热解重量分析,以在每次热处理和/或氢化处理之后确定每种材料的残余烃含量。样品Cr-100、Cr-150C-H2以及Cr-25C-H2的TGA(顶部迹线)以及DTA(底部迹线)光谱分别展示于图11、12以及13中。首先,样品Cr-100由于材料在分析期间的稍微氧化而在TGA实验开始时质量增加3%。质量然后在135℃与686℃之间稳定地降低,此时,材料保持其初始重量的78%。22%的质量损失可能归因于存在于材料中的烃((三甲基硅烷基)甲基或桥接亚烷基)的燃烧。样品Cr-100的DTA曲线展示位于322℃的尖锐放热峰以及位于470℃的较小放热峰。在150℃以及85巴H2下发生固态氢化之后,样品Cr-150C-H2展现存在于材料中的烃的量发生明显损失。TGA展示完全燃烧之后,材料的初始质量仅损失11.5%。DTA曲线显示位于316℃的较大、极宽放热峰以及位于506℃的较小放热峰。样品Cr-25C-H2保持其质量的91.5%。DTA曲线展示位于285℃的极宽放热峰以及位于576℃的较尖锐峰。剩余8.5wt%的烃无法通过在室温下进一步氢化24小时来除去。
对所有三种样品进行X射线光电子光谱分析且铬2p区域展示于图14-19中。对于图14-17中所示的样品Cr-100以及Cr-150C-H2来说,586.9以及587.3eV的较大2p1/2发射可以通过与Cr(OH)3的586.8eV的发射比较而指派给Cr(III)(参见Desimoni等人,《表面和界面分析(Surface and Interface Analysis)》,13,173,1998)。样品Cr-100以及Cr-150C-H2的模拟峰拟合还展示2p1/2区域中的位于584eV的较小发射,其可以指派给较低价Cr物质,因为铬金属的发射出现于583.5eV。不希望受理论所束缚,本发明人推测,样品Cr-100从Cr(IV)四烷基前驱物中发生的热沉淀可以促使氧化态还原为+3,表明除形成聚合物所必需的α-氢提取之外,还发生还原性Cr-C键均裂。样品Cr-100氢化而形成样品Cr-150C-H2之后,+3氧化态得以保持,表明氢不充当还原剂,但氢化物替换烃配位体。对于图18以及19中的样品Cr-25C-H2来说,在586.9eV观察到2p1/2区域中的较大发射,通过与Cr(OH)3比较,其可以指派给铬(III)物质。
样品Cr-100、Cr-150C-H2以及Cr-25C-H2的重量氢吸附-解吸附等温线展示于图20中。对于所有三种材料来说,在不饱和的情况下,在150巴下,等温线随着压力递增而线性升高。对于样品Cr-100来说,材料在150巴达到2.44wt%。吸附与解吸附等温线之间不存在滞后,展示氢吸附的可逆性。在150℃以及85巴H2下发生固态氢化之后,在不饱和的情况下,材料性能在150巴下稍微增加到3.1wt%。在150巴下,样品Cr-25C-H2达到最大值5.07wt%。虽然可以预计并入系统中存在一些性能损失,但此值非常接近于美国DOE的重量系统吸附目标5.5wt%。由于在150巴下未发现氢吸附饱和,因此有可能在高于150巴的压力下改进重量性能。为了确保准确度,使用碳AX-21作为标准物。
图21展示10个吸附-解吸附循环之后,Cr-25C-H2的PCT氢吸附-解吸附等温线。误差估算为±0.07wt%。Cr-25C-H2在0与150巴之间的循环重复吸附以及解吸附证明材料在10个循环中无活性损失。这是储氢材料商业化用于车辆应用中的重要特性。在150巴下10个循环的平均吸附值是5.12wt%。
利用高压拉曼光谱法研究样品Cr-25C-H2与氢的结合机制。图22展示样品Cr-25C-H2在50巴氢下以及空白样品架在50巴H2下的拉曼光谱的扩增区域。完整光谱展示于图23中。在空样品架与样品Cr-25C-H2的光谱中,在3568以及3807cm-1观察到归因于系统效应的信号。在样品Cr-25C-H2的光谱中仅观察到位于4075cm-1的信号且所述信号来源于物理吸附的H2。在两种光谱中,在约4128与4163cm-1之间还出现自由氢的Q-分支。计算表明,在3847cm-1与3936cm-1之间应该观察到库巴斯结合H2相对于铬(III)肼物质的H-H拉伸频率(参见Skipper等人,《物理化学杂志C》,116,19134,2002)。在样品Cr-25C-H2的光谱中,在氢气下,存在位于2789、2922以及3188cm-1的3个信号,其可以指派给库巴斯结合的H2。这些信号类似于针对氢化Ti(III)凝胶上的库巴斯结合H2所观察到的那些信号(参见Hoang等人,《材料化学(Chem.Mater.)》,25,4765,2013),但较宽,这可能归因于结合位点更分散于无定形的Cr材料中。还获得D2下的拉曼光谱。参见图23。最显著的是,Q-分支移到较低频率,如根据同位素效应所预计。位于2789以及3188cm-1的谱带不再存在,而位于2922cm-1的谱带现被Q-分支遮蔽。
在PCT吸附测量期间,添加氢的每次投料期间的样品Cr-25C-H2的量热曲线展示于图24中。与碳AX-21的放热量热曲线相比,从图24可以看出样品Cr-25C-H2的吸附过程是稍微吸热的,但氢第一次投料是放热的。第一次放热性投料可能起因于H2的物理吸附或归因于一些库巴斯结合位点的可用性。样品Cr-25C-H2的氢吸附焓是+0.37kJ mol-1H2。
实例2:氢化钒(IV)样品
合成
四苯基钒(IV)的制备
在室温下搅拌苯基锂(50mmol,25mL于二丁醚中的2.0M溶液)。然后通过注射器逐滴添加VCl4(2.03mL,12.5mmol)。反应混合物的颜色变为深褐色,温度升高且鼓泡相当剧烈。搅拌持续十五分钟,直到其已经停止鼓泡且已经冷却回到室温。然后过滤混合物,得到深褐色沉淀物以及褐色滤液。所得四苯基钒(IV)错合物不经进一步提纯即使用。
氢化钒(IV)的形成
紧接着将滤液转移到PARR不锈钢压力容器中且在惰性气氛下、在100℃下搅拌48小时。然后过滤混合物,得到黑色沉淀物。沉淀物在100℃下真空干燥四小时,得到228mg黑色精细粉末(V(IV)-100)。使黑色粉末在PARR容器中、在25℃下、在70巴压力下氢化48小时。所得材料然后在100℃下真空干燥4小时且允许冷却到室温,得到109.3mg黑色粉末(V(IV)-25C-H2)。
样品表征
样品V(IV)-100以及V(IV)-25C-H2的红外(IR)光谱展示于图25中。对于样品V(IV)-100来说,观察到位于2958cm-1、2919cm-1以及2868cm-1的C-H片段。在70巴下室温氢化之后,C-H片段的强度稍微降低(样品V(IV)-25C-H2),原因是烃配位体在氢解期间被氢化物替换。典型地,在1900±300cm-1区域的区域中观察到过渡金属-氢化物键,然而其强度可以是微弱的(参见Kaesz等人,《化学评论》,72,231,1972)。在此区域中存在位于1633cm-1的样品V(IV)-25C-H2片段。在KBr中,水显示位于3300以及1647cm-1的谱带,因此V(IV)-25C-H2光谱中位于1633cm-1的V-H片段可能被从手套箱到IR装置的快速转移步骤期间由KBr圆片吸附的水的O-H片段遮蔽。
在77K下所记录的样品V(IV)-100与V(IV)-25C-H2的氮吸附-解吸附等温线分别展示于图26和27中。两个样品均具有类型2等温线。样品V(IV)-100的BET表面积是0.6m2/g。氢化之后,样品V(IV)-25C-H2的表面积增加到2.2m2/g。表面积的增加可以归因于材料中烃的损失,从而在材料中产生新的路径。0P/Po与0.1P/Po之间不存在显著的斜率升高,表明两种材料中均不存在微孔隙。0.8与1.0P/Po之间的斜率升高起因于构造上的多孔性。
样品V(IV)-100的X射线粉末衍射(XRPD)图展示于图28中。材料基本上是无定形的。0-30°2θ区域中的少量反射可以归因于分析期间所用的玻璃毛细管。
氢吸附-解吸附研究
样品V(IV)-100以及V(IV)-25C-H2的重量氢吸附-解吸附等温线分别展示于图29和30中。在测试压力下,在不饱和的情况下,等温线随着压力递增而线性升高。对于样品V(IV)-100来说,材料在141巴下达到3.2wt%。在不完全可逆的情况下,等温线存在滞后。然而,在10-3托下施加真空五分钟完成解吸附,因此第二次吸附运作在141巴下达到3.2wt%。在100巴下,样品V(IV)-25C-H2达到最大值8.5wt%。这超过了美国DOE的重量吸附目标5.5wt%。为了确保准确度,使用碳AX-21作为标准物。
图31展示12个吸附-解吸附循环之后,V(IV)-25C-H2的PCT氢吸附-解吸附等温线。V(IV)-25C-H2在0与100巴之间的循环重复吸附以及解吸附证明材料在12个循环中无活性损失。在100巴下12个循环的平均吸附值是8.5wt%。
实例3:氢化锰(II)样品
合成
双(均三甲苯基)锰(II)的制备
氯化锰(II)(5.0043g,39.77mmol)与1,4-二噁烷(13.6mL,159.08mmol)一起在室温下搅拌1小时,得到浅粉红色糊状物。然后添加乙醚(50mL),得到浅粉红色悬浮液。在室温下,向其中逐滴添加均三甲苯基溴化镁(79.54mmol,79.54mL于乙醚中的1.0M溶液)且悬浮液从浅粉红色变为褐色。在室温下搅拌褐色悬浮液24小时,然后过滤,得到红褐色滤液以及浅米色沉淀物。用乙醚冲洗沉淀物三次。将洗涤液与滤液合并且真空浓缩,得到红褐色固体。然后将固体置于石油醚(100mL)中萃取且过滤,得到红褐色滤液以及浅褐色沉淀物。真空浓缩滤液,得到暗红褐色固体,在室温下真空干燥,得到红褐色粉末(1.95g,30%)。
氢化锰(II)的形成
双(均三甲苯基)锰(II)(1.9g,6.5mmol)在50mL温热的石油醚中搅拌,得到红褐色溶液。在惰性气氛下,将溶液转移到PARR不锈钢氢化容器中,在搅拌下,在100℃下加热48小时。过滤反应混合物,且所得深褐色沉淀物在100℃下真空干燥4小时,得到对空气湿气敏感的深褐色固体(Mn(II)-100)(120mg)。然后通过在室温下将粉末放置于PARR容器中且在100巴下对容器进行加压来使此材料与氢气发生反应。48小时之后,将材料在100℃下真空干燥四小时,得到深褐色精细粉末(Mn(II)-25C-H2)(58mg)。
样品表征
样品Mn(II)-100以及Mn(II)-25C-H2的红外(IR)光谱分别展示于图32和33中。对于样品Mn(II)-100来说,观察到位于2966cm1、2917cm-1和2857cm-1的C-H片段。在70巴下室温氢化之后,C-H片段的强度稍微降低(样品Mn(II)-25C-H2),原因是烃配位体在氢解期间被氢化物替换。在两种光谱中,还存在部分地被位于1640cm-1的水片段遮蔽的位于1740cm-1的片段,其属于过渡金属-氢化物片段的1900±300cm-1区域。参见例如Kaesz等人,《化学评论》,72,231,1972。
在77K下所记录的样品Mn(II)-100与Mn(II)-25C-H2的氮吸附-解吸附等温线分别展示于图34和35中。样品(II)-100展现6m2/g的BET表面积。室温氢化之后,样品Mn(II)-25C-H2的BET表面积降低到1.2m2/g。在两种样品中,吸附与解吸附等温线之间存在一些滞后,这意味着材料不是无孔材料。0与0.1P/Po之间存在非常小的斜率升高,表明不存在或存在非常少的微孔隙。样品Mn(II)-100在0.1与0.8P/Po之间适中的斜率升高起因于介孔隙且0.8与1.0P/Po之间的斜率升高起因于构造上的多孔性。
氢吸附-解吸附研究
样品Mn(II)-100以及Mn(II)-25C-H2的重量氢吸附-解吸附等温线分别展示于图36和37中。在不饱和的情况下,等温线随着压力递增而线性升高。对于样品Mn(II)-100来说,材料在130巴下达到2wt%且吸附与解吸附等温线之间存在一些小滞后。
在150巴下,样品Mn(II)-25C-H2达到最大值6.7wt%。这超过了美国DOE的重量吸附目标5.5wt%。因为在150巴下未发现氢吸附饱和,所以有可能在高于150巴的压力下改进重量性能。为了确保准确度,使用碳AX-21作为标准物。
实例4:氢化锰(II)样品
系列1
氯化锰(II)与含有2当量新戊基氯化镁的乙醚且与4当量的二噁烷一起搅拌,且在室温下搅拌溶液24小时。所得产物通过过滤分离且用100mL乙醚洗涤。真空除去溶剂且在40℃干燥产物1天(86%产率)。然后将所得产物溶解于40mL石油醚中,过滤且用100mL石油醚洗涤。真空除去溶剂且真空干燥产物4小时(24%产率)。然后将0.8g产物溶解于100mL石油醚中且在PARR容器中、在100℃下放置2天(不处在氢氛围下)。回收了188mg固体且真空干燥2小时,得到146mg固体(18.2%产率)。所得产物的红外(IR)光谱展示于图38中。
然后使产物在150℃和85巴H2下氢化4小时。将温度降低到100℃且真空干燥样品2小时。所得产物的红外(IR)光谱展示于图39中。图39的初始光谱之后2分钟和5分钟所获得的此相同样品的红外(IR)光谱分别展示于图40和41中。
如从图38可以看出,观察到位于1740cm-1的金属氢化物(Mn-H)片段。在氢处理后,此Mn-H片段的强度增长(图39),且在暴露于空气2分钟和5分钟后降低(图40和41)。不希望受理论所束缚,本发明人推测,图38中的Mn-H片段的存在(即,通过热沉淀形成、但在氢化之前的产物)表明已经发生σ-键复分解过程,其中与锰原子配位的新戊基配位体被正己基配位体(来自石油醚)替换,其然后经历β-氢化物消去而形成Mn-H键和1-己烯。此可能在当所用溶剂(石油醚)含有β-氢原子且容易发生C-H活化时发生。
如图39中所见,氢处理引起位于1740的Mn-H片段的强度增加且引起C-H片段的强度降低。如图40和41中所示,氢化产物暴露于空气导致羟基片段形成,其可能是水吸附至样品中和/或Mn-H键转化成Mn-OH键所引起。
系列2
氯化锰(II)与含有2当量新戊基氯化镁的乙醚且与4当量的二噁烷一起搅拌,且在室温下搅拌溶液24小时。所得产物通过过滤分离且用100mL乙醚洗涤。真空除去溶剂且在40℃干燥产物1天。然后将所得产物溶解于40mL石油醚中,过滤且用100mL石油醚洗涤。除去溶剂且真空干燥产物4小时。然后将0.8g产物溶解于100mL石油醚中且在PARR容器中、在298K、在73巴H2下放置2天。回收了193mg固体且真空干燥2小时,得到173mg固体(21.6%产率)。所得产物样品的红外(IR)光谱展示于图42中。
然后使产物在150℃和85巴H2下氢化4小时。将温度降低到100℃且真空干燥样品2小时。所得产物的红外(IR)光谱展示于图43中。图43的初始光谱之后5分钟所获得的此相同样品的红外(IR)光谱展示于图44中。
如可以从图42看出,观察到位于1740cm-1的金属氢化物(Mn-H)片段,表明系列1(在不氢化的情况下,使二烷基锰发生热沉淀)和系列2(利用氢化来形成二烷基锰)中所述的方法得到类似产物。
系列3
氯化锰(II)与含有2当量新戊基氯化镁的乙醚且与4当量的二噁烷一起搅拌,且在室温下搅拌溶液24小时。所得产物通过过滤来分离且用100mL乙醚洗涤。真空除去溶剂且在40℃干燥产物1天。然后将所得产物溶解于40mL石油醚中,过滤且用100mL石油醚洗涤。除去溶剂且真空干燥产物4小时。然后将0.8g产物溶解于100mL环己烷中且在PARR容器中、在100℃下放置2天(不处在氢氛围下)。回收了155mg固体且真空干燥2小时,得到148mg固体(18.5%产率)。所得产物样品的红外(IR)光谱展示于图45中。
然后使产物在150℃和85巴H2下氢化4小时。将温度降低到100℃且真空干燥样品2小时。所得产物的红外(IR)光谱展示于图46中。图46的初始光谱之后2分钟和5分钟所获得的此相同样品的红外(IR)光谱分别展示于图47和48中。
如从图45可以看出,使初始产物在环己烷(对C-H活化呈惰性的溶剂)中发生热沉淀,得到未展现位于1740cm-1的片段、但展现C-H片段的产物,表明在溶剂的C-H活化不存在下,可以发生涉及C-H活化(通过α-或γ-氢化物提取)来得到具有桥接烷基的有机金属聚合物的机制。图46展示产物的氢化引起C-H片段的强度降低和在1740cm-1出现Mn-H片段。如图47和48中所示,氢化产物暴露于空气导致位于1740cm-1的谱带的强度降低。
实例5:直接测量氢化锰(II)样品的H2吸附焓
双(新戊基)锰(II)是使用2014年6月13日所提交的美国专利申请第14/304,317号(作为美国公开案第2014/0370406号公开)中所述的二噁烷方法,由新戊基氯化镁制备,所述申请以全文引用的方式并入本文中。此化合物对空气非常敏感且自发地与甚至痕量的氧发生反应而形成绿色化合物,其存在不利地影响下游储氢性能。
(1.09g,5.53mmol)于100mL石油醚中搅拌,得到褐色溶液。将溶液放入PARR压力容器中且在氩气惰性气氛下、在100℃搅拌两天。在冷却到室温之后,过滤反应混合物,得到黑色沉淀物和无色滤液。黑色沉淀物在100℃真空干燥四小时,得到对空气湿气敏感的黑色粉末(105mg)(Mn-Cal-100)。然后通过将粉末放置于不锈钢样品架中来使此材料与氢气发生反应。将样品架附接到PCT-Pro仪器上。向样品中装入20巴H2且然后使样品架的温度提高到150℃。然后向样品中装入85巴H2。将样品冷却到100℃后,真空干燥样品2小时,随后冷却到室温,得到对空气湿气敏感的黑色粉末(53.7mg)(Mn-Cal-150-H2)。
样品表征
样品Mn-Cal-150-H2的红外(IR)光谱如下展示于图49中。由于烃仍然存留于材料中,所以位于2964cm-1的片段对应于C-H片段。位于1021cm-1和1094cm-1的片段起因于C-O且可能归因于乙醚从前驱物Mn(新戊基)2的合成转移到最终材料。还存在位于1646cm-1的片段,其可能归因于将KBr圆片从手套箱快速转移到IR光谱仪的期间,所述圆片所拾取的大气中的水。
氢吸附研究
样品Mn-Cal-150-H2的重量氢吸附-解吸附等温线展示于图50中。材料在90巴和40℃下达到重量氢吸附最大值4.47wt%。如图50中所示,等温线随着压力递增(直到90巴,其中氢吸附达到平稳阶段)而线性升高。从图49可以看出,材料中仍然存在剩余的烃。除非已经发生氧化,否则在通过进一步氢化去除烃后,预计储氢性能超过4.47wt%。
量热法
样品Mn-Cal-150-H2的量热曲线如下展示于图51中。为了测定热流(焦耳),在特定压力下投予氢气期间对应于热流变化的每个峰是使用Setaram供应的用于C80量热器的Calisto数据处理软件进行积分。为了获得100巴下的总热量(焦耳),将每次投氢的热量(焦耳)加在一起,得到0.513J。为了解释当氢引入熔炉中的C80池时气体升温的效应,从空白测量值中减去100巴下的热量(0.513J-0.625J=-0.112J)。然后通过将热量(焦耳)除以所吸附氢的摩尔数来计算H2吸附焓。在这个实例中,样品Mn-Cal-150-H2的氢吸附焓是-0.11kJ mol-1H2。
通过库巴斯相互作用储存氢的材料的氢吸附焓典型地是-20-40kJ mol-1H2。因为通过直接测量所得到的样品Mn-Cal-150-H2的值位于这个范围外部且接近于零,所以可能涉及第二种过程,其可以归因于材料的扭转而产生新的库巴斯氢结合位点。
实例6:氢化铬(II)样品的合成
CrCl2(THF)1.13的制备
氯化铬(II)(5.353g,43.55mmol)于80mL THF中搅拌,得到绿色悬浮液。然后在回流下搅拌悬浮液72小时。然后真空除去过量THF且将由此获得的浅绿色固体在50℃下真空干燥3小时,得到浅绿色粉末(8.8914g)。
双[(三甲基硅烷基)甲基]铬(II)的制备
向CrCl2(THF)1.13(5.1095g,25mmol)于200mL 40-60℃石油醚中的搅拌悬浮液中添加(三甲基硅烷基)甲基锂溶液(50mmol,50mL于戊烷中的1.0M溶液)。浆液颜色紧接着变为深褐色。在室温下搅拌混合物12小时,然后过滤,且用石油醚(3×20mL)洗涤残余物。将深褐色滤液浓缩且在室温下真空干燥48小时,得到深褐色固体(5g,89%产率)。还参见Schulzke等人,《有机金属化合物》,21,3810,2002。
氢化铬(II)样品的制备
双[(三甲基硅烷基)甲基]铬(II)(5g,22.1mmol)于250mL 30-60°石油醚中搅拌,得到深褐色溶液。将溶液放入PARR压力容器中且容器在10巴H2下加压72小时。然后将压力提高到70巴维持48小时。然后将容器在100℃下、在氩气气氛下加热72小时,随后将反应物在100℃和80巴H2下加热72小时。过滤反应物,得到黑色沉淀物以及黑色滤液。用石油醚(3×10mL)冲洗沉淀物。然后将沉淀物在100℃下真空干燥4小时,得到对空气湿气敏感的黑色粉末Cr(II)-100(0.3155g)。
样品表征
样品Cr(II)-100的红外(IR)光谱展示于图52中。位于2950cm-1的片段可以归因于C-H片段,其来源于尚未从材料中除去的三甲基硅烷基配位体。位于1261cm-1的片段来源于存在于三甲基硅烷基中的C-Si。存在宽C-O片段,其可以归因于存在于来自前驱物的材料中的残余THF。存在位于1664cm-1的中等强度吸光度,其接近于针对基质分离CrH2所观察到的Cr-H片段。然而,在KBr中,水显示位于3300和1647cm-1的片段。因此,位于1664cm-1的片段有可能来源于KBr圆片所拾取的大气中的水且Cr-H片段被O-H片段遮蔽。
对样品Cr(II)-100进行X射线光电子光谱分析,以测定存在于材料中的铬的氧化态。Cr 2p区域展示于图53中。峰拟合展示存在三种不同的促进Cr 2p3/2发射的氧化状态。位于576.4eV的较大发射可以归因于Cr(II),原因是其类似于双(苯)铬的575.4eV发射(参见Pignataro等人,《化学物理学快报(Chemical Physics Letters)》,20,35,1973)。位于577.3eV的发射可以归因于Cr(III),原因是其接近于CrCl3的577.3eV发射。位于579.4eV的不太强的发射可以归因于Cr(IV),原因是其与Cr2(CrO4)3中的Cr(IV)的位于579.7eV的发射位于相同区域中(参见Volkov等人,《俄罗斯无机化学杂志(Zhurnal Neorganicheskoi Khimii)》,39,877,1994)。
氢吸附研究
样品Cr(II)-100的重量氢吸附(底部迹线)-解吸附(顶部迹线)等温线展示于图54中。在不饱和的情况下,在170巴,等温线随着压力递增而线性升高。材料在170巴和298K下具有0.92wt%的重量储氢能力。吸附等温线与解吸附等温线之间存在一些滞后。此材料的性能低于材料Cr-100,其在150巴和298K下达到2.44wt%。两种材料之间的差异可能在于,烷基铬前驱物是使用不同的合成程序制得。在此实例中,依循Gambarotta所报道的路线(参见Gambarotta等人,《有机金属化合物》,221,3810,2002),所述路线始于CrCl2(THF)1.13和两当量的烷基锂。由此产生深褐色Cr(II)烷基。使Cr(II)烷基发生氢化,得到储氢特性比深紫色烷基铬(IV)四聚物错合物分解所产生的材料更差的材料。
因此,始于深紫色烷基铬(IV)(如实例1中)得到储氢材料Cr-100(其中烷基铬(IV)的分解引起还原,得到Cr(III)物质)。始于烷基铬(II)前驱物(如实例6中)得到具有混合氧化态(其中存在Cr(II)、Cr(III)和(Cr(IV))的材料。混合价的Cr材料(Cr(II)-100)的储氢能力低于样品Cr-100。
实例7:氢化锰(II)样品的替代合成
用研杵和研钵将2.96g(23.5mmol)MnCl2(预先经亚硫酰氯干燥)研磨成精细粉末且在50mL乙醚中搅拌。添加新戊基氯化镁(47mmol,于20mL乙醚中)且搅拌反应混合物20分钟。然后历经5小时逐滴添加8.01mL(94mmol)二噁烷且反应物颜色从粉红色变为灰色/绿色,同时其变稠。然后添加20mL额外乙醚且在室温下搅拌反应物24小时。然后过滤反应物,得到褐色/绿色滤液和白色沉淀物。用3×30mL乙醚洗涤沉淀物(滤液从深褐色变成浅橙色)。过滤后,真空除去溶剂,得到深褐色固体,随后在45℃真空干燥2小时,得到3.7g浅褐色固体(81%)。然后将固体溶解于40mL石油醚中,搅拌20分钟且过滤,得到白色沉淀物和褐色滤液。真空除去溶剂,得到2.925g褐色固体(62%)。使此固体在石油醚中、在100℃热沉淀48小时,得到900mg精细黑色固体。
此氢化锰(II)样品的红外(IR)光谱展示于图55中。
实例8:氢化钒(IV)样品的替代合成
本实例描述一种制备烷基钒(IV)前驱物(四均三甲苯基钒(IV))的改进方法,其使用格林纳试剂(均三甲苯基MgBr),从而避免使用昂贵的锂试剂。
合成
四均三甲苯基钒(IV)的制备
四氯化钒(2g,10.38mmol)于乙醚(40mL)中在25℃搅拌,得到深红色溶液。然后逐滴添加均三甲苯基溴化镁(41.52mL于乙醚中的1.0M溶液)。添加格林纳试剂后,反应混合物冒泡且从深红色变成黑色。搅拌24小时之后,过滤反应混合物,得到灰色沉淀物(2.1g)和黑色滤液。真空浓缩滤液,得到黑色油状物。用石油醚(50mL)萃取油状物,得到褐色沉淀物和深红色/紫色滤液。将滤液真空浓缩且干燥,得到深红色/紫色油状物(4.6g,84%)。
热沉淀
将2.3g(4.35mmol)四均三甲苯基钒(IV)于50mL石油醚中搅拌,得到深褐色溶液。然后在氩气气氛下将溶液放入PARR不锈钢压力容器中。容器在100℃下搅拌48小时。冷却到室温之后,过滤反应混合物,得到深褐色滤液和黑色沉淀物。将黑色沉淀物在100℃下真空干燥4小时,得到对空气敏感的深褐色固体V(Mes)-100(360mg)。
氢化钒(IV)样品的制备
然后使对空气敏感的褐色固体V(Mes)-100在100巴H2和25℃下、在溶剂不存在下发生氢化,得到氢化钒样品V(Mes)-100H2。
样品表征
样品V(Mes)-100(底部迹线)和V(Mes)-100H2(顶部迹线)的红外(IR)光谱展示于图56中。对于样品V(mes)-100片段来说,观察到位于2917以及2968cm-1的C-H片段。在110巴下室温氢化之后,C-H片段的强度稍微降低(样品V(Mes)-100H2),原因是烃配位体在氢解期间被氢化物替换。典型地,在1900±300cm-1区域的区域中观察到过渡金属-氢化物键,然而其强度可以是微弱的(参见Kaesz等人,《化学评论》,72,231-281,1972)。对于样品V(Mes)-110H2来说,观察到此区域中位于1630cm-1的片段和位于1625cm-1的肩峰。在KBr中,水显示位于3300和1647cm-1的谱带。V(Mes)-110H2光谱中位于1625cm-1的肩峰(其可归因于V-H片段)可能稍微被在从手套箱到IR装置的快速转移步骤期间由KBr圆片吸附的水的位于1630cm-1的O-H片段遮蔽。
氢吸附-解吸附研究
样品V(Mes)-100(底部迹线,吸附(方形)、解吸附(三角形))和V(Mes)-100H2(顶部迹线,吸附(方形)、解吸附(三角形))的重量氢吸附-解吸附等温线展示于图57中。
在测试压力下,在不饱和的情况下,等温线随着压力递增而线性升高。对于样品V(Mes)-100来说,材料在110巴下达到1.3wt%。等温线中不存在滞后,表明不存在显著的动力学阻障要克服以使氢完全解吸附。室温氢化之后,样品V(Mes)-100的重量显著降低且储氢性能提高。样品V(Mes)-110H2在110巴下达到最大值1.7wt%。为了确保准确度,使用碳AX-21作为标准物。
本发明范围不受本文所述的特定实施例限制。实际上,除了本文中所述的那些内容之外,所属领域的技术人员根据前述说明和附图将显而易知本发明的各种修改。希望这些修改属于所附权利要求书的范围内。
本申请中引用专利、专利申请、公开案、产品说明书和方案,其揭露内容是以全文引用的方式并入本文中用于所有目的。
- 还没有人留言评论。精彩留言会获得点赞!