确定待测样本的SMN1基因是否存在七号外显子缺失的方法和系统与流程
本发明涉及生物信息领域,具体地,本发明涉及确定待测样本的smn1基因是否存在七号外显子缺失的方法和系统。
背景技术:
脊髓性肌萎缩症(sma)是一种常染色体隐性遗传疾病,为一组可起病于婴儿期,儿童期或青少年期的疾病,其特征是由脊髓前角细胞与脑干内运动核进行性变性引起的骨骼肌萎缩,患者智力不受影响。临床主要表现为下运动神经元进行性对称性肌无力和肌萎缩,近端重于远端,下肢重于上肢。该病发病为1/6000~1/10000,居致死性常染色体隐性遗传病第二位,目前尚无有效治疗方法。在中国的携带率为1/62,在世界范围内携带率1/30~1/40。
两个高度同源运动神经元存活基因:smn1基因和smn2基因,被认为与脊髓型肌肉萎缩症相关,这两个基因相似性高达99%。其中smn1基因是其功能的主要决定者,其同源性缺失或突变引起脊髓型肌肉萎缩症,而smn2基因的拷贝数与发病的严重程度相关。smn1基因含有9个外显子(exon1,2a,2b,and3-8),编码294个氨基酸残基,组成38kd的smn蛋白,该蛋白的功能尚未完全清晰,但对于正常的运动神经元是十分必要的。而由于smn2基因剪切方式上存在缺陷,大多数smn2基因产生的premrna会产生可变剪切,产生一个截断的,功能缺失的蛋白。在smn2的产物中,仅15%是正常的、有功能的smn蛋白。目前,sma分子诊断的情况显示,约95%~98%的脊髓型肌肉萎缩症患病个体为smn1基因7号外显子缺失或截断的纯合突变。因此,临床上可以通过检测smn1基因7号外显子的缺失情况对脊髓型肌肉萎缩症患者进行快速、简便的基因诊断。
目前主流的基因检测方法为目标区域捕获+高通量测序(ngs),该技术可以实现同时对多个样本多个基因进行捕获及测序,这对于包含多种基因和疾病的筛查产品,可有效降低成本和交付周期。脊髓型肌肉萎缩症作为一种高携带率疾病,扩展性携带者筛查指南中明确指出,脊髓型肌肉萎缩症为需筛查疾病。由于smn1基因和smn2基因的高度同源性,使得利用这两个基因的高通量测序数据存在一定困难。目前多家公司推出的筛查产品,均采用补充方法,如定量pcr或者多重连接探针扩增技术(mlpa)等对脊髓型肌肉萎缩症进行检测,这额外增加了筛查产品的成本。虽有个别文献报道利用目标区域捕获+高通量测序检测smn1基因7号外显子缺失的方法,由于文献方法存在缺陷,市场上还没有成熟产品应用此方法。
技术实现要素:
本申请是基于发明人对以下事实和问题的发现和认识做出的:
2015年larson等人发表名为“validationofahighresolutionngsmethodfordetectingspinalmuscularatrophycarriersamongphase3participantsinthe1000genomesproject”的文章,提出了应用高通量测序数据对smn1的7号外显子缺失检测的方法(larsonetal.bmcmedicalgenetics(2015)16:100.doi10.1186/s12881-015-0246-2)。文章中应用批次内的所有样本来选取控制基因集,并计算smn基因相对控制基因集的权重均值(后称放缩系数)。目的是以批次内所有样本在smn1、smn2基因及控制基因集上的深度的平均表现作为正常样本的表现情况,来判断每一个样本的拷贝数。但发明人发现,文章是基于在筛查样本中,正常样本(smn1与smn2均为2拷贝的样本)占多数的情况来考虑的,而对于异常情况(即smn1、smn2的拷贝数存在缺失或重复的样本占比较大时),利用上述文章的方法检测出现错误的风险会很大
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,针对上述问题,本申请引入了控制样本集的概念,即首先选出smn1与smn2均是2拷贝的样本做参考。在有相同实验条件的历史检测样本数据时,可直接选取smn1与smn2均是2拷贝的样本。否则,在批次内选取smn1的深度占smn1加smn2深度的比例(通过覆盖特异位点的reads数计算得到)在0.43到0.57之间的样本作为控制样本集。经测试,这一方法的引入可以有效解决批次内总样本数少或阳性样本过多时,检测结果中灰区样本(无法判断的样本)很多,或出现假阴、假阳样本的问题。
在本发明的第一方面,本发明提出了一种确定待测样本的smn1基因是否存在七号外显子缺失的方法。根据本发明的实施例,所述方法包括:(1)对来自于总样本集的多个核酸样本分别进行测序,所述总样本集包括待测试样本和至少一个控制样本,所述多个核酸样本均含有smn编码基因和至少一个控制基因,所述smn编码基因包括:smn1七号外显子编码序列;smn2七号外显子编码序列;smn1七号外显子左侧和右侧毗邻区编码序列;和smn2七号外显子左侧和右侧毗邻区编码序列;(2)针对所述总样本集中的每一个样本,基于步骤(1)的测序结果,选择来源于所述smn编码基因和所述至少一个控制基因的测序读段;(3)针对所述待测试样本,确定smn1七号外显子参数,所述smn1七号外显子参数与来源于所述smn1七号外显子编码序列的测序读段数目呈正相关;(4)针对所述待测试样本,基于所述至少一个控制样本中所述至少一个控制基因的测序读段,对所述smn1七号外显子参数进行矫正;(5)基于经过矫正的所述smn1七号外显子参数,预测来源于所述smn编码基因的测序读段归属于smn1七号外显子编码序列的概率;以及(6)基于所述概率,确定所述待测样本的smn1基因是否存在七号外显子缺失。根据本发明实施例的方法在对smn1基因的7号外显子缺失检测上,其灵敏度、特异度相比现有技术有明显提升,并可有效区分出杂合缺失与纯合缺失的样本;当批次内正常样本(smn1基因和smn2基因的7号外显子各为2拷贝)占较比小时,通过选取控制样本集大幅提高了检测精度。
根据本发明的实施例,上述方法还可以进一步包括如下附加技术特征至少之一:
根据本发明的实施例,所述smn1七号外显子编码序列包含第一突变位点,所述第一突变位点位于chr5:70247773;所述smn1七号外显子左侧毗邻区编码序列包括第二突变位点,所述第二突变位于chr5:70247724;所述smn1七号外显子右侧毗邻区编码序列包括第三突变位点,所述第三突变位于chr5:70247921;所述smn2七号外显子编码序列包含第四突变位点,所述第四突变位点位于chr5:69372353;所述smn2七号外显子左侧毗邻区编码序列包括第五突变位点,所述第五突变位于chr5:69372304;以及所述smn2七号外显子右侧毗邻区编码序列包括第六突变位点,所述第六突变位于chr5:69372501。
根据本发明的实施例,在步骤(3)中,针对所述待测样本,所述smn1七号外显子参数是通过下列步骤确定的:(3-1)基于所述待测样本的测序结果,分别确定携带所述第一至第六突变位点的所述测序读段的数目;(3-2)基于步骤(3-1)中所得到的所述第一至第六突变位点的所述测序读段的数目,确定第一至第三比例,其中,所述第一比例y=b/b,其中,b表示携带所述第一突变位点的所述测序读段的数目,b表示来自于携带所述第一或第四突变位点的所述测序读段的数目,所述第二比例x=a/a,其中,a表示携带所述第二突变位点的所述测序读段的数目,a表示来自于携带所述第二或第五突变位点的所述测序读段的数目,和所述第三比例x=m/m,其中,m表示携带所述第三突变位点的所述测序读段的数目,m表示来自于携带所述第三或第六突变位点的所述测序读段的数目;(3-3)基于所述第一至第三比例,按照下列公式,确定参数r和r,其中,r构成所述smn1七号外显子参数:当所述第一比例与所述第二比例的差异绝对值以及所述第一比例和所述第三比例的差异绝对值至少之一超过0.1时,r=b,r=b;当所述第一比例与所述第二比例的差异绝对值以及所述第一比例和所述第三比例的差异绝对值均不超过0.1时,r=a+b+m,r=a+b+m。
根据本发明的实施例,所述方法进一步包括:(3-4)基于所述参数r,确定所述待测样本是否合格。
根据本发明的具体实施例,所述参数r小于200,是所述待测样本不合格的指示。
根据本发明的具体实施例,所述方法进一步包括:(3-5a)基于所述参数r和r,确定第四比例q,其中所述第四比例q=r/r;(3-5b)判断所述控制样本是否合格,其中,所述第四比例q在0.43~0.57范围内是所述控制样本合格的指示;或者基于所述第四比例q在0.43~0.57范围内,初步确定所述待测样本的smn1基因不存在七号外显子缺失。
根据本发明的实施例,所述至少一个控制基因是通过下列步骤确定的:(a)基于所述至少一个控制样本的测序结果,选择多个候选基因,所述多个候选基因在至少一部分所述控制样本中的测序深度高于预定阈值;(b)针对所述多个候选基因的每一个,分别在所述至少一个控制样本的每一个中,计算第五比例zk,i=si/hk,i,其中,k表示候选基因编号,i表示所述样本的编号,si表示第i号样本中smn基因的测序深度,hk,j表示在所述第i号样本中第k号候选基因的测序深度;以及(c)基于所述第五比例,确定所述至少一个控制基因。
根据本发明的实施例,在步骤(c)中,所述控制基因满足下列标准的至少之一:(c-1)在所述至少一个控制样本之间,所述控制基因的测序深度的变异系数是最小的前10位;和(c-2)在所述至少一个控制样本之间,所述第五比例的变异系数是最小的前10位。
根据本发明的实施例,所述预定阈值是通过如下方式确定的:基于所述至少一个控制样本的测序结果,所述样本全部基因的至少一部分的测序深度按照从小到大的顺序进行排列;以及基于所述排列结果,确定所述预定阈值,所述阈值为不小于处于5%位置的基因所对应的测序深度。
根据本发明的具体实施例,所述阈值为处于5%位置的基因所对应的测序深度。
根据本发明的实施例,在所述至少一个控制样本的至少90%中,所述候选基因的测序深度大于所述预定阈值。换句话说,也就是候选基因的测序深度在至少一个控制样本中小于预定阈值的比例不大于10%。
根据本发明的实施例,在步骤(4)中,所述矫正是通过所述smn1七号外显子参数乘以矫正系数进行的,其中,所述矫正系数是通过下列公式确定的:
其中,zk表示在所述待测样本中smn基因的测序深度与所述第k编号基因的测序深度的比例,k表示所述控制基因集中的所述控制基因的总数目,
根据本发明的再一具体实施例,
根据本发明的实施例,当通过公式
根据本发明的实施例,在步骤(5)中,经过矫正的smn1的七号外显子参数所对应的七号外显子编码序列的测序读段数服从二项分布,应用贝叶斯模型计算来源于所述smn编码基因的测序读段归属于smn1七号外显子编码序列的概率pi。具体地,矫正后的七号外显子编码序列的测序读段数服从二项分布ri'=θiri~bin(ri,pi),这里pi表示矫正后的比对到smn的测序读段数实际来自smn1的概率,ri'表示经过矫正的smn1的七号外显子编码序列的测序读段数,ri表示未经矫正地smn1的七号外显子编码序列的测序读段数,ri表示比对到smn基因的7号外显子的测序读段数总数。由于beta分布为二项分布的共轭先验分布的密度函数,假设先验分布pi~beta(1,1),则后验分布为pi~beta(1+ri',1+ri)。
根据本发明的再一实施例,在步骤(6)中,基于所述pi的95%置信区间[a’,b’],确定所述待测样本的smn1基因是否存在七号外显子缺失,其中,a’大于0.38是所述待测样本的smn1基因不存在七号外显子缺失的指示,b’小于0.38是所述待测样本的smn1基因存在七号外显子缺失的指示;a’不大于0.38且b’不小于0.38无法判断待测样本是否存在七号外显子缺失。具体地,考虑smn1为1拷贝,smn2为2拷贝的情形,此时pi的理论值应为1/3(其余缺失情形均小于1/3);允许一类错误0.05,最终设定阈值为0.38;进而通过pi的95%置信区间[a,b]与0.38的关系判断smn1的7号外显子缺失情况,即当a’>0.38时,为阴性(不存在七号外显子缺失);b’<0.38时,为阳性(存在七号外显子缺失的指示);a’<=0.38且0.38<=b’时,为灰区,即无法判断。
根据本发明的实施例,当所述待测样本的smn1基因存在七号外显子缺失时,所述方法进一步包括通过公式
根据本发明的实施例,smn1基因拷贝数是0是smn1基因7号外显子纯合缺失的指示;smn1基因拷贝数不小于1是smn1基因7号外显子杂合缺失的指示;smn1基因拷贝数在0~1之间是smn1基因7号外显子灰区缺失的指示。
相比于现有技术仅对smn1是否缺失做判断,但无法判断smn1及smn2的拷贝数的缺陷,根据本发明实施例的方法,可计算出smn1与smn2的拷贝数,并可用以区分杂合缺失与纯合缺失的样本,进而可以进一步统计人群中smn1、smn2拷贝数,为研究脊髓性肌萎缩症(sma)这种常染色体隐性遗传疾病奠定了基础。
在本发明的第二方面,本发明提出了一种确定待测样本的smn1基因是否存在七号外显子缺失的系统。根据本发明的实施例,所述系统包括:测序装置,所述测序装置用于对来自于总样本集的多个核酸样本分别进行测序,所述总样本集包括待测试样本和至少一个控制样本,所述多个核酸样本均含有smn编码基因和至少一个控制基因,所述smn编码基因包括:smn1七号外显子编码序列;smn2七号外显子编码序列;smn1七号外显子左侧和右侧毗邻区编码序列;和smn2七号外显子左侧和右侧毗邻区编码序列;选择smn编码基因和控制基因的装置,所述选择smn编码基因和控制基因的装置与所述测序装置相连,用于针对所述总样本集中的每一个样本,基于所述测序装置的测序结果,选择来源于所述smn编码基因和所述至少一个控制基因的测序读段;确定smn1七号外显子参数装置,所确定smn1七号外显子参数装置与所述选择smn编码基因和控制基因的装置相连,用于述针对所述待测试样本,确定smn1七号外显子参数,所述smn1七号外显子参数与来源于所述smn1七号外显子编码序列的测序读段数目呈正相关;矫正装置,所述矫正装置与所述确定smn1七号外显子参数装置相连,用于针对所述待测试样本,基于所述至少一个控制样本中所述至少一个控制基因的测序读段,对所述smn1七号外显子参数进行矫正;预测归属装置,所述预测归属装置与所述矫正装置相连,用于基于经过矫正的所述smn1七号外显子参数,预测来源于所述smn编码基因的测序读段归属于smn1七号外显子编码序列的概率;以及确定装置,所述确定装置与所述预测归属装置相连,用于基于所述概率,确定所述待测样本的smn1基因是否存在七号外显子缺失。根据本发明实施例的系统在对smn1基因的7号外显子缺失检测上,其灵敏度、特异度相比现有技术有明显提升,并可有效区分出杂合缺失与纯合缺失的样本;当批次内正常样本(smn1基因和smn2基因的7号外显子各为2拷贝)占较比小时,通过选取控制样本集大幅提高了检测精度。
根据本发明实施例的上述系统适于执行上述的根据本发明实施例的确定待测样本的smn1基因是否存在七号外显子缺失的方法,其优势、效果以及附加技术特征如前所述,在此不再赘述。
附图说明
图1是根据本发明实施例的确定待测样本的smn1基因是否存在七号外显子缺失的系统的结构示意图;以及
图2是根据本发明另一实施例的确定待测样本的smn1基因是否存在七号外显子缺失的系统的结构示意图。
具体实施方式
下面将结合附图对根据本发明实施例的确定待测样本的smn1基因是否存在七号外显子缺失的系统做进一步详细描述。可以理解的是,下面结合附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
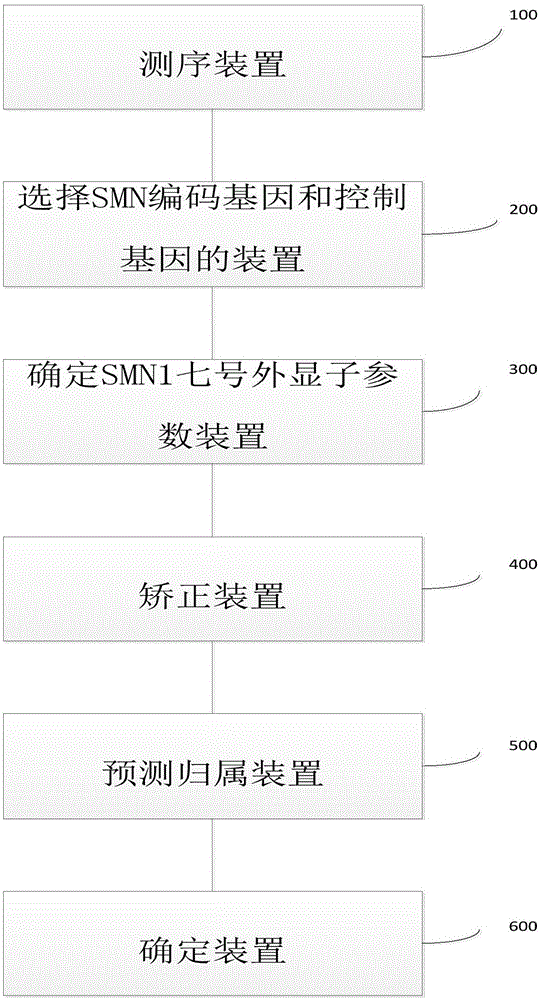
本发明提出了一种确定待测样本的smn1基因是否存在七号外显子缺失的系统。根据本发明的实施例,参考图1,所述系统包括:
测序装置100,所述测序装置用于对来自于总样本集的多个核酸样本分别进行测序,所述总样本集包括待测试样本和至少一个控制样本,所述多个核酸样本均含有smn编码基因和至少一个控制基因,所述smn编码基因包括:
smn1七号外显子编码序列,所述smn1七号外显子编码序列包含第一突变位点,所述第一突变位点位于chr5:70247773;
smn2七号外显子编码序列,所述smn2七号外显子编码序列包含第四突变位点,所述第四突变位点位于chr5:69372353;
smn1七号外显子左侧和右侧毗邻区编码序列,所述smn1七号外显子左侧毗邻区编码序列包括第二突变位点,所述第二突变位于chr5:70247724,所述smn1七号外显子右侧毗邻区编码序列包括第三突变位点,所述第三突变位于chr5:70247921;和
smn2七号外显子左侧和右侧毗邻区编码序列,所述smn2七号外显子左侧毗邻区编码序列包括第五突变位点,所述第五突变位于chr5:69372304,所述smn2七号外显子右侧毗邻区编码序列包括第六突变位点,所述第六突变位于chr5:69372501;
选择smn编码基因和控制基因的装置200,所述选择smn编码基因和控制基因的装置200与所述测序装置100相连,用于针对所述总样本集中的每一个样本,基于所述测序装置的测序结果,选择来源于所述smn编码基因和所述至少一个控制基因的测序读段;
确定smn1七号外显子参数装置300,所确定smn1七号外显子参数装置300与所述选择smn编码基因和控制基因的装置相连200,用于述针对所述待测试样本,确定smn1七号外显子参数,所述smn1七号外显子参数与来源于所述smn1七号外显子编码序列的测序读段数目呈正相关,
所述确定smn1七号外显子参数装置300适于执行以下操作:
(3-1)基于所述待测样本的测序结果,分别确定携带所述第一至第六突变位点的所述测序读段的数目;
(3-2)基于步骤(3-1)中所得到的所述第一至第六突变位点的所述测序读段的数目,确定第一至第三比例,其中,
所述第一比例y=b/b,其中,b表示携带所述第一突变位点的所述测序读段的数目,b表示来自于携带所述第一或第四突变位点的所述测序读段的数目,
所述第二比例x=a/a,其中,a表示携带所述第二突变位点的所述测序读段的数目,a表示来自于携带所述第二或第五突变位点的所述测序读段的数目,和
所述第三比例x=m/m,其中,m表示携带所述第三突变位点的所述测序读段的数目,m表示来自于携带所述第三或第六突变位点的所述测序读段的数目;
(3-3)基于所述第一至第三比例,按照下列公式,确定参数r和r,其中,r构成所述smn1七号外显子参数:
当所述第一比例与所述第二比例的差异绝对值以及所述第一比例和所述第三比例的差异绝对值至少之一超过0.1时,r=b,r=b;
当所述第一比例与所述第二比例的差异绝对值以及所述第一比例和所述第三比例的差异绝对值均不超过0.1时,r=a+b+m,r=a+b+m;
(3-4)基于所述参数r,确定所述待测样本是否合格,其中,所述参数r小于200,是所述待测样本不合格的指示;
(3-5a)基于所述参数r和r,确定第四比例q,其中所述第四比例q=r/r;
(3-5b)判断所述控制样本是否合格,其中,所述第四比例q在0.43~0.57范围内是所述控制样本合格的指示;或者
基于所述第四比例q在0.43~0.57范围内,初步确定所述待测样本的smn1基因不存在七号外显子缺失;
矫正装置400,所述矫正装置400与所述确定smn1七号外显子参数装置300相连,用于针对所述待测试样本,基于所述至少一个控制样本中所述至少一个控制基因的测序读段,对所述smn1七号外显子参数进行矫正;
预测归属装置500,所述预测归属装置500与所述矫正装置400相连,用于基于经过矫正的所述smn1七号外显子参数,预测来源于所述smn编码基因的测序读段归属于smn1七号外显子编码序列的概率,
所述预测归属装置500适于执行以下操作:经过矫正的smn1的七号外显子参数所对应的七号外显子编码序列的测序读段数服从二项分布,应用贝叶斯模型计算来源于所述smn编码基因的测序读段归属于smn1七号外显子编码序列的概率;以及
确定装置600,所述确定装置600与所述预测归属装置500相连,用于基于所述概率,确定所述待测样本的smn1基因是否存在七号外显子缺失,
所述确定装置600适于执行以下操作:
基于所述pi的95%置信区间[a’,b’],确定所述待测样本的smn1基因是否存在七号外显子缺失,
其中,a’大于0.38是所述待测样本的smn1基因不存在七号外显子缺失的指示,b’小于0.38是所述待测样本的smn1基因存在七号外显子缺失的指示;a’不大于0.38且b’不小于0.38无法判断待测样本是否存在七号外显子缺失。
根据本发明的具体实施例,所述至少一个控制基因是通过下列步骤确定的:
(a)基于所述至少一个控制样本的测序结果,选择多个候选基因,所述多个候选基因在至少一部分所述控制样本中的测序深度高于预定阈值;
(b)针对所述多个候选基因的每一个,分别在所述至少一个控制样本的每一个中,计算第五比例zk,i=si/hk,i,其中,k表示候选基因编号,i表示所述样本的编号,si表示第i号样本中smn基因的测序深度,hk,j表示在所述第i号样本中第k号候选基因的测序深度;以及
(c)基于所述第五比例,确定所述至少一个控制基因,所述控制基因满足下列标准的至少之一:
(c-1)在所述至少一个控制样本之间,所述控制基因的测序深度的变异系数是最小的前10位;和
(c-2)在所述至少一个控制样本之间,所述第五比例的变异系数是最小的前10位。
根据本发明的具体实施例,所述预定阈值是通过如下方式确定的:基于所述至少一个控制样本的测序结果,所述样本全部基因的至少一部分的测序深度按照从小到大的顺序进行排列;以及基于所述排列结果,确定所述预定阈值,所述阈值为不小于处于5%位置的基因所对应的测序深度;
根据本发明的再一具体实施例,所述阈值为处于5%位置的基因所对应的测序深度。
根据本发明的具体实施例,在所述至少一个控制样本的至少90%中,所述候选基因的测序深度大于所述预定阈值。换句话说,也就是候选基因的测序深度在至少一个控制样本中小于预定阈值的比例不大于10%。
根据本发明的具体实施例,所述矫正装置适于执行以下操作,所述矫正通过所述smn1七号外显子参数乘以矫正系数进行,其中,所述矫正系数是通过下列公式确定的:
其中,
zk表示在所述待测样本中smn基因的测序深度与所述第k编号基因的测序深度的比例,
k表示所述控制基因集中的所述控制基因的总数目,
根据本发明的具体实施例,
n表示控制样本基因中样本的总数,i表示样本编号,k表示基因编号。
根据本发明的具体实施例,当通过公式
根据本发明的再一具体实施例,参考图2,所述系统进一步包括确定smn1基因的拷贝数装置700,所述确定smn1基因的拷贝数装置700与所述确定装置600相连,所述确定smn1基因的拷贝数装置700适于执行以下操作:
当所述待测样本的smn1基因存在七号外显子缺失时,
通过公式
其中,c1,i或c2,i不大于0.1,是smn1基因或smn2基因拷贝数是0的指示,
c1,i或c2,i大于0.1但小于0.5,是smn1基因或smn2基因拷贝数在0~1之间的指示,
c1,i或c2,i不小于0.5但小于1.485,是smn1基因或smn2基因拷贝数是1的指示,
c1,i或c2,i不小于1.485但小于2.324,是smn1基因或smn2基因拷贝数是2的指示,
c1,i或c2,i不小于2.324但小于2.743,是smn1基因或smn2基因拷贝数在2~3之间的指示,
c1,i或c2,i不小于2.743,是smn1基因或smn2基因拷贝数不低于3的指示。
根据本发明的具体实施例,smn1基因拷贝数是0是smn1基因7号外显子纯合缺失的指示;smn1基因拷贝数不小于1是smn1基因7号外显子杂合缺失的指示;smn1基因拷贝数在0~1之间是smn1基因7号外显子灰区缺失的指示。
根据本发明实施例的系统适于执行根据本发明实施例的确定待测样本的smn1基因是否存在七号外显子缺失的方法,在对smn1基因的7号外显子缺失检测上,其灵敏度、特异度相比现有技术有明显提升,并可有效区分出杂合缺失与纯合缺失的样本;当批次内正常样本(smn1基因和smn2基因的7号外显子各为2拷贝)占较比小时,通过选取控制样本集大幅提高了检测精度。
下面结合具体实施例进一步阐明本发明确定待测样本的smn1基因是否存在七号外显子缺失的方法。下面描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
应用ngs下机数据估算smn1/2基因7号外显子的拷贝数的分析流程如下:
实施例1原始下机数据处理
步骤a,将下机数据应用过滤软件(soapnuke软件,版本:1.5.2)过滤掉低质量的测序读段(reads)。
步骤b,将reads使用比对软件(bwa软件,版本:0.7.12)比对到人参考基因组(hg19),使用picard(一种基本序列处理软件,版本:1.87)标记重复序列,应用gatk(一种用于二代重测序数据分析的软件,版本:3.2)进行重比对和碱基质量值校正。
步骤c,应用gatk的depthofcoverage工具获取捕获区域的reads覆盖信息。需要输出位点reads数信息文件及区域平均覆盖度文件(后缀为.sample_interval_summary文件)。
实施例2估算smn1/2基因的7号外显子拷贝数
步骤a,计算比对到smn1基因和smn2基因的reads数。smn1基因与smn2基因在7号外显子区域序列仅有1个位点差异(smn1上的chr5:70247773;smn2上的chr5:69372353,记为位点b),在7号外显子两侧的内含子各有1个位点差异(smn1基因上的chr5:70247724和smn2基因上的chr5:69372304记为位点a,smn1基因上chr5:70247921和smn2基因上的chr5:69372501,记为位点c)。假设批次共n个样本,计算位点b处smn1基因reads数占smn总reads数的比值yi=bi/bi,其中bi为第i个样本中比对到smn1基因的b位点的reads数,bi为比对到smn总reads数。同样对于位点a、c,计算xi=ai/ai、zi=ci/ci。记ri为比对到smn1的7号外显子的reads数,ri为比对到smn基因的7号外显子的reads总数,若|xi-yi|>0.1或|zi-yi|>0.1,则ri=bi,ri=bi;否则ri=ai+bi+ci,ri=ai+bi+ci。计算比对到smn1的reads占smn总reads的比例πi=ri/ri。此外设定ri<200的样本为不合格样本。
步骤b,控制样本集的选取。批次内选取0.43≤πi≤0.57的样本(通常为smn1基因和smn2基因的7号外显子各为2拷贝的样本)作为控制样本集。如有相同实验条件的历史数据,也可选取smn1基因和smn2基因的7号外显子检测结果均为2拷贝的样本,作为控制样本集。
步骤c,控制基因集的选取。在深度足够(在至少10%的样本中低于所有样本所有基因的第5百分位数)的基因里,计算zk,i=si/hk,i,其中si为smn基因的平均深度,hk,i为第k个基因的平均深度。按照下面两个条件来选取控制基因集(1)在控制样本集中平均深度变化较小(2)在控制样本集中zk,i变化较小。其中变化程度应用变异系数cv进行衡量。选取两个条件下cv值较小的前10个基因,取并集作为控制基因集。
步骤d,计算smn基因相对控制基因集的权重均值
pi|ri',pi~beta(α+ri',ri-ri'+β),
通过后验分布的累计分布函数计算p(pi≤0.38|ri',ri)作为第i个样本为smn1基因7号外显子缺失携带者的概率。计算pi的95%置信区间。若置信区间完全大于0.38,样本为阳性;完全小于0.38,样本为阴性;0.38在置信区间内,则判为灰区。
步骤e,计算smn1基因的初始拷贝数
这里0.5代表0拷贝到1拷贝之间但无法细分的情况。2.5代表2拷贝到3拷贝之间但无法细分的情况。3代表有3拷贝或更多拷贝数的情况。
样本的smn1基因7号外显子缺失的最终判定结果,以步骤d的结果为准。步骤d判断为阳性时,则需结合步骤e的结果进行纯合缺失与杂合缺失的判断。若步骤e中smn1基因的预测拷贝数n=0,则判断样本为纯合缺失,若n=0.5,则判断样本为缺失灰区样本,若n≥1,则判断样本为杂合缺失。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
- 该技术已申请专利。仅供学习研究,如用于商业用途,请联系技术所有人。
- 技术研发人员:郭凤禹;宋立洁;孙隽;王亚玲;范林林;彭智宇
- 技术所有人:天津华大医学检验所有限公司;广州华大基因医学检验所有限公司;深圳华大基因股份有限公司;深圳华大临床检验中心
- 我是此专利的发明人
- 该领域下的技术专家
- 如您需求助技术专家,请点此查看客服电话进行咨询。
- 1、薛老师:1.CRISPR-Cas系统 2.基因编辑 3.基因修复 4.天然产物合成 5.单分子技术开发与应用
- 2、张老师:1.探索新型氧化还原酶结构-功能关系,电催化反应机制 2.酶电催化导向的酶分子改造 3.纳米材料、生物功能多肽对酶-电极体系的影响4. 生物电化学传感和生物电合成体系的设计与应用。
- 3、豆老师:1.环境纳米材料及挥发性有机化合物(VOCs) 2.CO污染物的催化氧化 3.低温等离子体 4.吸脱附等控制技术
- 4、赵老师:1.高分子材料改性及加工技术 2.微孔及过滤材料 3.环境友好高分子材料
- 5、邬老师:1.高分子材料的共混与复合 2.涉及材料功能化及结构与性能的研究; 高分子热稳定剂的研发
- 如您是高校老师,可以点此联系我们加入专家库。
- 还没有人留言评论。精彩留言会获得点赞!