一种芳香乙胺及其衍生物的制备方法
1.本发明涉及有机合成制备化学领域,尤其涉及一种芳香乙胺及其衍生物的制备方法。
背景技术:
2.芳香乙胺是有机合成和医药工业重要的中间体之一。例如苯乙胺是驱虫药物吡喹酮和治疗糖尿病药物苯乙双胍合成的原料。胡椒乙胺可以用于合成黄连素。此外3,4-二甲氧基苯乙胺是合成多种药物如甲基多巴胺酚,治疗关节炎的延胡索乙素的中间体,是合成许多异喹啉生物碱,如罂粟碱、海罂粟碱、维尼碱等的中间体,也可以作为合成抗高血压药物,支气管平滑肌迟缓剂,胃分泌抑制剂等的原料。3,4-二甲氧基苯乙胺作为一种重要的有机化工原料,可以用于合成新一代受体阻断剂,同时也可以用于合成抗癌药物苯并蒽嗪甲酰胺衍生物。
3.芳香乙胺具有广泛的应用价值,目前国内外市场对其需求逐年增长。其合成方法有大量文献报道,主要分为三类:(1)硝基化合物还原法;(2)hofmann酰胺降解法;(3)芳基乙腈加氢还原法,其中最常用的途径是以取代的苯甲醛为原料与硝基甲烷缩合生成β-硝基苯乙烯,再经氢化铝锂还原的方法。此方法的还原剂氢化铝锂价格昂贵,应用于大规模的工业化生产会产生较高成本。使用苯甲酰胺水解制备芳香乙胺具有经济和操作简便等优点,但目前仍缺少高效经济合成苯甲酰胺的方法。有鉴于此,特提出本发明。
技术实现要素:
4.本发明的第一目的是提供一种苯甲酰胺的制备方法,该方法使用廉价金属催化的高效、高纯度、高收率的苯甲酰胺的制备方法,该方法操作简单,耗费时间短,工艺流程大大缩减,原料易得,适合工业生产。
5.本发明的第二目的是提供一种芳香乙胺及其衍生物的制备方法,该方法可快速的将苯甲酰胺制备成芳香乙胺及其衍生物,满足现有的芳香乙胺及其衍生物的需求。
6.为了实现上述目的,本发明特采用以下技术方案:
7.本发明提供了一种苯甲酰胺的制备方法,其包括如下步骤:
8.n-乙烯基苯甲酰胺和芳基硼酸发生反应后制备得到苯甲酰胺;
9.其中,所述n-乙烯基苯甲酰胺与所述芳基硼酸的摩尔比1.0:(2.5-3.5);
10.所述芳基硼酸的化学结构式为:
11.其中,r为-cl,-br,-f,-i,-nph2,-cn,-come,-cooet,-ome,-ocf3,-nhboc,-cf3,-sime3,-sme,-ch2oh,-cho,-come或烷基中的一种或几种的组合,ar为芳香环或芳香杂环,包括如下基团的一种:
[0012][0013]
所述芳基硼酸是如下结构的一种:
[0014][0015]
[0016]
优选地,所述r为-ome,所述ar为所述芳基硼酸的结构为
[0017]
当然了,在进行制备苯甲酰胺的反应过程中,还添加有锰催化剂,添加剂,碱和溶剂,所述n-乙烯基苯甲酰胺与所述锰催化剂、所述添加剂、所述碱、所述溶剂的摩尔比为1.0:(0.025-0.1):(4.0-6.0):(0.1-1.0):(35-70)。
[0018]
优选地,所述n-乙烯基苯甲酰胺与所述芳基硼酸、所述锰催化剂、所述添加剂、所述碱、所述溶剂的摩尔比为1:(2.7-3.2):(0.03-0.09):(4.5-5.5):(0.3-0.7):(40-65)。
[0019]
优选地,所述n-乙烯基苯甲酰胺与所述芳基硼酸、所述锰催化剂、所述添加剂、所述碱、所述溶剂的摩尔比为1:3:0.05:5.0:0.5:42。
[0020]
优选地,在添加的原料中,所述锰催化剂的选择为mn2(co)8br2和mn(co)5br的至少一种,所述添加剂为水和乙醇的至少一种,所述碱为碳酸钾和醋酸钠的至少一种,所述溶剂为甲基叔丁基醚,乙醚。1,2-二氯乙烷,四氢呋喃中的一种或几种的组合。
[0021]
更优选地,所述锰催化剂为mn2(co)8br2,所述添加剂为水,所述碱为碳酸钾,所述溶剂为甲基叔丁基醚。
[0022]
优选地,发生所述反应的过程中,所述反应的温度为100-130℃,所述反应的时间为0.20-2h。
[0023]
优选地,所述发生反应时,加温至120℃,所述反应的时间为1h。
[0024]
除此之外,反应的温度还可以为101℃、104℃、105℃、107℃、108℃、109℃、110℃、112℃、114℃、115℃、117℃、118℃、119℃、122℃、125℃、127℃、129℃等等,所述反应的时间还可以为0.22h、0.27h、0.3h、0.4h、0.7h、1.3h、1.9h等等。
[0025]
本发明可以实现的主要原因是锰催化的反应机制。在上述反应条件下,锰催化剂与芳基硼酸发生转金属化作用生成活性芳基锰催化物种。随后n-乙烯基苯甲酰胺上的碳碳双键迁移插入至碳锰键中,所得中间体经质子化脱去锰催化剂可得到目标产物苯甲酰胺。本发明加入物料的摩尔比是有限制的,少于或大于指定摩尔比范围均会使反应产率和效率降低。
[0026]
本发明使用了商业可得的芳基硼酸作为芳基的来源,可供选择的范围十分广泛。芳基硼酸上芳环的种类和取代基可以根据合成需要改变,从而制备一系列结构复杂的苯甲酰胺。然而传统合成苯甲酰胺或芳香乙胺及其衍生物的方法中,由于反应条件剧烈苛刻,很多敏感官能团不能在反应中兼容,所以只能制备结构简单的产物,底物适应性较为狭窄。
[0027]
本发明使用了新型的锰催化剂,采用一种全新的催化机制。锰催化剂与芳基硼酸发生转金属化并与n-乙烯基苯甲酰胺发生迁移插入的步骤在体系中各物料相互配合下可快速进行,使反应时间大大缩短。并且该反应直接一步对烯基进行官能团化修饰,减少了反应步骤,提升了合成效率。
[0028]
本发明还提供了一种芳香乙胺及其衍生物的制备方法,其是由上述制备方法制得的苯甲酰胺与氢氧化钠反应制得的。
[0029]
优选地,所述苯甲酰胺与所述氢氧化钠的摩尔比为1.0:(4.0-10.0);
[0030]
优选地,所述苯甲酰胺与所述氢氧化钠的摩尔比为1.0:5.0。
[0031]
通过实践发现,上述所涉及的参数,比如原料之间的摩尔比、反应温度、反应时间等均需要控制在比较适宜的范围内,不能过高也不能过低,因为如果反应温度、反应时间过长一方面不够经济,也会使得反应中所涉及到的诸多物料不能保证在最好的活性条件下进行反应,温度太低,反应时间太短又会出现原料大量剩余的现象,降低所要得到的目标产物的产率。
[0032]
本发明所制备的芳香乙胺的化学结构式为:
[0033]
其中,r为-cl,-br,-f,-i,-nph2,-cn,-come,-cooet,-ome,-ocf3,-nhboc,-cf3,-sime3,-sme,-ch2oh,-cho,-come或烷基中的一种或几种的组合,ar为芳香环或芳香杂环,包括如下基团的一种:
[0034][0035]
本发明与现有技术相比,至少有以下优异之处:
[0036]
(1)本发明芳基硼酸与n-乙烯基苯甲酰胺在锰催化下反应得到n-芳香乙基苯甲酰胺,提供了一种全新的制备苯甲酰胺的方法,反应步骤少,操作方法简单,原料易得,且具有高效率,高收率,高纯度等特点,适合工业生产。
[0037]
(2)本发明通过利用n-芳香乙基苯甲酰胺和氢氧化钠水解后制备芳香乙胺及其衍生物,能够满足现有的芳香乙胺及其衍生物的使用,并且步骤减少,反应时间短,能更快速的制备出芳香乙胺及其衍生物满足需求。
附图说明
[0038]
通过阅读下文优选实施方式的详细描述,各种其他的优点和益处对于本领域普通技术人员将变得清楚明了。附图仅用于示出优选实施方式的目的,而并不认为是对本发明的限制。而且在整个附图中,用相同的参考符号表示相同的部件。在附图中:
[0039]
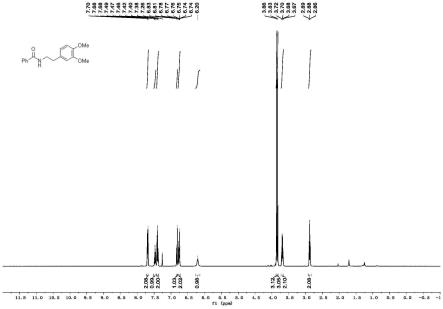
图1为本发明实施例所提供的制备方法制备出的苯甲酰胺的核磁共振氢谱图;
[0040]
图2为本发明实施例所提供的制备方法制备出的苯甲酰胺的核磁共振碳谱图;
[0041]
图3为本发明实施例所提供的制备方法制备出的芳香乙胺的核磁共振氢谱图;
[0042]
图4为本发明实施例所提供的制备方法制备出的芳香乙胺的核磁共振碳谱图。
具体实施方式
[0043]
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会
理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0044]
实施例1
[0045]
1)制备苯甲酰胺按照如下步骤操作:
[0046]
在100ml的厚壁耐压瓶中加入特氟龙外壳的磁力搅拌子,加入n-乙烯基苯甲酰胺1.18g,芳基硼酸4.37g,锰催化剂196mg,添加剂0.72g,碱0.55g和溶剂40ml,加温后发生氢芳基化反应,反应结束后的产物经过分离,得到苯甲酰胺纯品。
[0047]
在本发明中,将芳基硼酸选择为3,4-二甲氧基苯硼酸,将锰催化剂选择为mn2(co)8br2,添加剂选择为水,碱选择为碳酸钾,溶剂选择为甲基叔丁基醚,经过柱色谱分离,得到的苯甲酰胺为n-(3,4-二甲氧基苯乙基)苯甲酰胺,产率为89%。
[0048]
在上述制备方法中,根据上述的各个组分的用量,可以得出n-乙烯基苯甲酰胺与芳基硼酸、锰催化剂、添加剂、碱、溶剂的摩尔比为1:3:0.05:5.0:0.5:42;
[0049]
所采用的芳基硼硼酸的结构式为
[0050]
本实施例为了达到更好的效果,将加温的温度选择加热到120℃,将加温反应的时间选择为1h。
[0051]
2)制备芳香乙胺按照如下步骤进行操作:
[0052]
采用上述步骤1)的制备方法所制备出来的苯甲酰胺为原料进行制备芳香乙胺:
[0053]
在反应器中加入n-(3,4-二甲氧基苯乙基)苯甲酰胺和氢氧化钠,并加入乙醇作为溶剂,加温后反应,反应结束后经乙酸乙酯萃取得到3,4-二甲氧基苯乙胺,即得到芳香乙胺,产率为92%,目标产物总产率为82%。
[0054]
其中,加温的温度为100℃,加温的时间选择为10h。
[0055]
将本实施例制备得到的n-(3,4-二甲氧基苯乙基)苯甲酰胺和3,4-二甲氧基苯乙胺的结构采用核磁共振仪器、高分辨质谱仪进行确认。
[0056]
所得到的n-(3,4-二甲氧基苯乙基)苯甲酰胺的化学结构式为:
[0057][0058]
具体表征数据如下:
[0059]
如图1所示:1hnmr(400mhz,cdcl3)δ7.73
–
7.66(m,2h),7.47(t,j=7.2hz,1h),7.40(t,j=7.6hz,2h),6.82(d,j=8.0hz,1h),6.79
–
6.73(m,2h),6.20(s,1h),3.86(s,3h),3.83(s,3h),3.69(q,j=6.4hz,2h),2.88(t,j=6.8hz,2h)。氢谱与预期产物图谱结构一致。
[0060]
如图2所示:
13
cnmr(100mhz,cdcl3)δ167.4,149.1,147.7,134.6,131.4,131.4,128.5,126.8,120.7,111.9,111.4,55.9,55.8,41.2,35.2.碳谱与预期产物图谱结构一致。
[0061]
高分辨质谱的具体表征数据为:hrms m/z(esi)calcd for c
17h19
no3(m+h)
+
:286.1438;found:286.1425。高分辨质谱与预期产物图谱结构一致。
[0062]
所得到的3,4-二甲氧基苯乙胺的化学结构式为:
[0063][0064]
具体表征数据如下:
[0065]
如图3所示:1hnmr(500mhz,cdcl3)δ6.78
–
6.61(m,3h),3.87
–
3.70(m,6h),2.94
–
2.80(m,2h),2.69
–
2.54(m,2h),1.17(s,2h)。氢谱与预期产物图谱结构一致。
[0066]
如图4所示:
13
cnmr(125mhz,cdcl3)δ148.6,147.2,132.2,120.5,111.8,111.1,55.6,55.5,43.4,39.4。碳谱与预期产物图谱结构一致。
[0067]
高分辨质谱的具体表征数据为:hrms m/z(esi)calcd for c
10h15
no2(m+h)
+
:182.1176;found:182.1171.。高分辨质谱与预期产物图谱结构一致。
[0068]
实施例2
[0069]
1)制备苯甲酰胺按照如下步骤操作:
[0070]
在100ml的厚壁耐压瓶中加入特氟龙外壳的磁力搅拌子,加入n-乙烯基苯甲酰胺1.19g,芳基硼酸4.37g,锰催化剂196mg,添加剂0.72g,碱0.55g和溶剂40ml,加温后发生氢芳基化反应,反应结束后的产物经过分离,得到苯甲酰胺纯品。
[0071]
在本发明中,将芳基硼酸选择为3,4-二甲氧基苯硼酸,将锰催化剂选择为mn(co)5br,添加剂选择为乙醇,碱选择为醋酸钠,溶剂选择为乙醚,经过柱色谱分离,得到的苯甲酰胺为n-(3,4-二甲氧基苯乙基)苯甲酰胺,产率为87%。
[0072]
在上述制备方法中,根据上述的各个组分的用量,可以得出n-乙烯基苯甲酰胺与芳基硼酸、锰催化剂、添加剂、碱、溶剂的摩尔比为1:3:0.05:5.0:0.5:42;
[0073]
所采用的芳基硼硼酸的结构式为
[0074]
本实施例为了达到更好的效果,将加温的温度选择加热到130℃,将加温反应的时间选择为0.5h。
[0075]
2)制备芳香乙胺按照如下步骤进行操作:
[0076]
采用上述步骤1)的制备方法所制备出来的苯甲酰胺为原料进行制备芳香乙胺:
[0077]
在反应器中加入n-(3,4-二甲氧基苯乙基)苯甲酰胺和氢氧化钠,并加入乙醇作为溶剂,加温后反应,反应结束后经乙酸乙酯萃取得到3,4-二甲氧基苯乙胺,即得到芳香乙胺,产率为90%,目标产物总产率为80%。
[0078]
其中,加温的温度为100℃,加温的时间选择为10h。
[0079]
将本实施例制备得到的n-(3,4-二甲氧基苯乙基)苯甲酰胺和3,4-二甲氧基苯乙胺的结构采用核磁共振仪器、高分辨质谱仪进行确认,所得到的相应化合物的化学结构式与实施例1一致。
[0080]
实施例3
[0081]
1)制备苯甲酰胺按照如下步骤操作:
[0082]
在100ml的厚壁耐压瓶中加入特氟龙外壳的磁力搅拌子,加入n-乙烯基苯甲酰胺1.18g,芳基硼酸4.37g,锰催化剂196mg,添加剂1.84g,碱0.55g和溶剂50ml,加温后发生氢芳基化反应,反应结束后的产物经过分离,得到苯甲酰胺纯品。
[0083]
在本发明中,将芳基硼酸选择为3,4-二甲氧基苯硼酸,将锰催化剂选择为mn2(co)8br2,添加剂选择为乙醇,碱选择为碳酸钾,溶剂选择为四氢呋喃和1,2-二氯乙烷按体积比为1:1混合而成,经过柱色谱分离,得到的苯甲酰胺为n-(3,4-二甲氧基苯乙基)苯甲酰胺,产率为88%。
[0084]
在上述制备方法中,根据上述的各个组分的用量,可以的出n-乙烯基苯甲酰胺与芳基硼酸、锰催化剂、添加剂、碱、溶剂的摩尔比为1:3:0.05:5.0:0.5:50;
[0085]
所采用的芳基硼硼酸的结构式为
[0086]
本实施例为了达到更好的效果,将加温的温度选择加热到100℃,将加温反应的时间选择为1.5h。
[0087]
2)制备芳香乙胺按照如下步骤进行操作:
[0088]
采用上述步骤1)的制备方法所制备出来的苯甲酰胺为原料进行制备芳香乙胺:
[0089]
在反应器中加入n-(3,4-二甲氧基苯乙基)苯甲酰胺和氢氧化钠,并加入乙醇作为溶剂,加温后反应,反应结束后经乙酸乙酯萃取得到3,4-二甲氧基苯乙胺,即得到芳香乙胺,产率为92%,目标产物总产率为82%。
[0090]
其中,加温的温度为100℃,加温的时间选择为10h。
[0091]
将本实施例制备得到的n-(3,4-二甲氧基苯乙基)苯甲酰胺和3,4-二甲氧基苯乙胺的结构采用核磁共振仪器、高分辨质谱仪进行确认,所得到的相应化合物的化学结构式与实施例1一致。
[0092]
实施例4-7
[0093]
具体实施方式与实施例1一致,不同之处在于改变了制备苯甲酰胺和芳香乙胺时的各个组分之间的摩尔配比与芳基硼酸的结构式,具体如表1、表2、表3所示。
[0094]
实施例8-9
[0095]
具体实施方式与实施例1一致,不同之处在于改变了制备苯甲酰胺时的各个组分之间的摩尔比,具体如表1所示。
[0096]
实施例10-12
[0097]
具体实施方式与实施例1一致,不同之处在于改变了芳基硼酸的结构式,具体如下表3所示。
[0098]
比较例1
[0099]
具体实施方式与实施例1一致,不同之处在于提高了n-乙烯基苯甲酰胺的用量,具体如下表1所示。
[0100]
比较例2
[0101]
具体实施方式与实施例1一致,不同之处在于提高了芳基硼酸的用量,具体如下表
1所示。
[0102]
比较例3
[0103]
具体实施方式与实施例1一致,不同之处在于提高了锰催化剂的用量,具体如下表1所示。
[0104]
比较例4
[0105]
具体实施方式与实施例1一致,不同之处在于降低了碱的用量,具体如下表1所示。
[0106]
表1各组分摩尔比对苯甲酰胺产率的影响
[0107][0108][0109]
表2各组分摩尔比对芳香乙胺产率的影响
[0110] 苯甲酰胺氢氧化钠产率
实施例414.088%实施例5110.091%实施例618.090%实施例785.082%
[0111]
表3芳基硼酸结构式对苯甲酰胺产率的影响
[0112][0113][0114]
通过比较分析实施例1-12和对比例1-4的实验结果,可知实施例1所述的n-乙烯基苯甲酰胺与所述芳基硼酸、所述锰催化剂、所述添加剂、所述碱、所述溶剂的摩尔比为最优,即1:3:0.05:5.0:0.5:42。最优的物料摩尔比是经过多次实验才得出的,实际操作时改变其中任意条件均不能得到最佳结果。如比较例1中,降低了芳基硼酸的摩尔比,导致产率有较为明显的下降。比较例2中提高了芳基硼酸的摩尔比,产率并没有明显上升,同时造成了反应物料的大量浪费。比较例3中提高了锰催化剂的摩尔比也不能促进反应产率的提升,还会造成催化剂的浪费,并非最经济的选择。在比较例2和比较例3中,芳基硼酸与锰催化剂的摩尔比相较于最优条件偏离过大或过小均会导致反应产率的降低,这个是由于改变芳基硼酸
与锰催化剂的摩尔比会使芳基硼酸与锰催化剂转金属化的速率和后续生成的活性锰物种对底物迁移插入的速率不匹配,进而导致产率的降低。在比较例4中,可以看出碱的摩尔比对反应产率也有较大影响,碱的量低于最优范围仅能获得60%的收率,与最优的实施例1差距较大,这是由于碱起到了促进芳基硼酸和锰催化剂之间的转金属化形成活性芳基锰物种的作用,碱加入的量过少会导致关键的转金属化步骤较难发生,进而使得反应产率降低。本发明还考察了芳基硼酸的适用范围,较为复杂的芳基硼酸在本发明的条件下也能够反应,如含有敏感官能团或杂芳环等的芳基硼酸,进而能够合成一系列结构复杂的苯甲酰胺或芳香乙胺。这些复杂苯甲酰胺或芳香乙胺的合成在传统合成路线中很难实现,体现了本发明良好的底物兼容性,为合成复杂结构的苯甲酰胺或芳香乙胺提供了一条简便的合成路径。
[0115]
最后,可以理解的是,以上实施方式仅仅是为了说明本发明的原理而采用的示例性实施方式,然而本发明并不局限于此。对于本领域普通技术人员而言,在不脱离本发明的原理和实质的情况下,可以做出各种变型和改进,这些变型和改进也视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1