一种唑啉草酯及其中间体的制备方法与流程
1.本发明属于化学合成领域,具体涉及一种唑啉草酯及其中间体的制备方法。
背景技术:
2.唑啉草酯是一种具有选择性、内吸传导性的芽后型禾本科杂草除草剂,能有效防除小麦及大麦田中的一年生禾本科杂草,非常适用于春季谷物。其对野燕麦、黑麦草、狗尾草、硬草、茼草、日本看麦娘、棒头草等禾本科杂草有良好防效功能,尤其对一些恶性禾本科杂草如野燕麦、黑麦草、硬草和茼草的防效接近100%。其具有高效、广谱、安全性高、施药适期宽、耐雨水冲刷等特点,并且在动物、植物、土壤和环境中降解很快,不滞留、不蓄积,也不会淋溶到地下水中。与其他麦田除草剂相比,唑啉草酯有着巨大的发展潜力和市场竞争力。
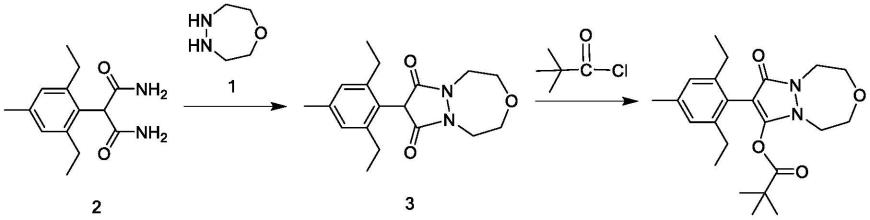
3.唑啉草酯的合成方法一般是通过化合物2和化合物1反应,合成化合物3,为了节省工艺步骤,化合物3通常不经纯化处理,直接与特戊酰氯反应,合成化合物4唑啉草酯。所以,化合物3是唑啉草酯合成过程中非常重要的中间体。
[0004][0005]
cn108794505a和cn108264492a均公开了化合物3的制备方法,两者均是将化合物2和化合物1一次性加入至反应容器中,加热回流,进行反应,收率分别为84%和90%。上述两种方法的收率均有待提高,且cn108264492a还使用了大量的三乙胺,三乙胺毒性较高、后处理繁琐,同时也增加了工艺成本。
[0006]
cn106928253a也公开了化合物3的制备方法:将化合物2、化合物1的氢溴酸盐和三乙胺一次性加入至反应容器中,加热反应,收率更低,仅有76%。该方法同样使用了大量的三乙胺(4.2eq),同样存在毒性高、后处理繁琐、工艺成本高的问题。
技术实现要素:
[0007]
发明要解决的问题
[0008][0009]
发明人发现,在化合物3的合成过程中常常会产生异构胺杂质和脱羧杂质,这两种杂质会在化合物3和特戊酰氯的反应过程中再分别生成新的杂质,这两种新的杂质在后处理过程中难以除去,从而影响唑啉草酯的产品收率和纯度。因此,在化合物2与化合物1的反应过程中,有必要控制异构胺杂质和脱羧杂质的相对含量。
[0010]
然而,现有技术均未研究化合物3的制备工艺对异构胺杂质和脱羧杂质含量的影响。因此,亟待开发出一种反应转化率和收率高、杂质含量低(尤其是异构胺杂质和脱羧杂质)、原料的原子利用率高、毒性低且后处理简便(例如,不使用三乙胺等试剂)的制备唑啉草酯及其中间体的工艺方法。
[0011]
本发明旨在提供一种唑啉草酯及其中间体(即化合物3)的制备方法,该工艺方法能够降低异构胺杂质和脱羧杂质的含量、反应转化率和收率高、原子利用率高、不使用三乙胺等有机碱。
[0012]
用于解决问题的方案
[0013]
为了解决上述问题,本发明提供了一种唑啉草酯中间体的制备方法,其包括以下步骤:
[0014][0015]
将化合物1和与水不互溶的有机溶剂混合,加热至100-140℃,加入化合物2,进行反应,得到化合物3;所述反应还包括以下条件中的至少一个:
[0016]
(1)反应压力为0.06-0.09mpa;
[0017]
(2)所述化合物2的加料方式为分批加料。
[0018]
优选地,在所述分批加料的过程中,所述化合物2的每批次加料量与所述化合物1的总加料量的摩尔比为0.1-0.5:1,更优选0.2-0.4:1,进一步优选0.25:1。
[0019]
优选地,所述化合物1的总加料量与所述化合物2的总加料量的摩尔比为1-5:1,更优选1-1.2:1,进一步优选1.05:1。
[0020]
进一步地,在所述分批加料的过程中,每批次加料的间隔时间为10-90分钟,优选30-75分钟,更优选60分钟。
[0021]
优选地,所述反应压力为0.065-0.085mpa,更优选0.067-0.082mpa。
[0022]
优选地,所述与水不互溶的有机溶剂为芳香类溶剂,优选氯苯、二甲苯或二氯苯,进一步优选氯苯。
[0023]
优选地,所述与水不互溶的有机溶剂与所述化合物2的总加料量的摩尔比为2:1以上。
[0024]
优选地,所述加热的温度为110-130℃,更优选118-125℃。
[0025]
优选地,所述制备方法还包括后处理的步骤。
[0026]
优选地,所述后处理的步骤包括:反应结束后降温,加入盐酸水溶液,分相,取有机相,即得化合物3;或者
[0027]
所述后处理的步骤包括:反应结束后降温,加入盐酸,过滤,即得化合物3;进一步地,所述降温的温度为20℃以下,所述盐酸与所述化合物2的总加料量的摩尔比为1。
[0028]
另外,本发明还提供了一种唑啉草酯的制备方法,其包括以下步骤:
[0029][0030]
其中,化合物3由上述唑啉草酯中间体的制备方法得到。
[0031]
发明的效果
[0032]
本发明对唑啉草酯中间体(即化合物3)的制备工艺进行了改进,通过先加热反应液,后分批加入化合物2的方式,显著缩短了反应时间,有效避免了脱羧杂质的大量产生;通过微负压的方式,降低反应的温度,减少氨气在反应中的存留时间,从而减少了异构胺杂质的产生。最终,本发明的工艺方法可以将异构胺杂质和脱羧杂质的含量有效控制至较低水平,保证了终产品唑啉草酯的质量,避免了复杂的除杂工艺,大大减低了工艺成本;并且提高了化合物2的转化率和化合物1的原子利用率,提高了反应收率,缩短了反应时间;同时反应过程中也未使用三乙胺等有机碱,毒性低,避免了复杂的后处理和回收步骤,适合工业化放大生产。
具体实施方式
[0033]
在本发明中,“唑啉草酯中间体”指化合物3(或其酮式-烯醇式互变异构体)。在化合物3的制备工艺中,发明人发现了异构胺杂质和脱羧杂质两种杂质,其含量大小会直接影响下一步合成唑啉草酯的反应,并且影响终产品唑啉草酯的品质。现有技术中,基本上没有对上述两种杂质进行研究,且现有工艺的合成转化率和收率不高,使用的三乙胺毒性较大,后处理和回收工艺繁琐。
[0034][0035]
本发明对化合物3的制备工艺进行了改进,通过先加热反应液,后分批加入化合物2的方式,显著缩短了反应时间,有效避免了脱羧杂质的大量产生;通过微负压的方式,降低反应的温度,减少氨气在反应中的存留时间,从而减少了异构胺杂质的产生。
[0036]
具体地,本发明提供的化合物3的制备方法包括以下步骤:将化合物1和与水不互溶的有机溶剂混合,加热至100-140℃,加入化合物2,进行反应,得到化合物3;其中,所述反应还包括以下条件中的至少一个:
[0037]
(1)反应压力为0.06-0.09mpa;
[0038]
(2)所述化合物2的加料方式为分批加料。
[0039]
在一些实施方式中,在所述分批加料的过程中,所述化合物2的每批次加料量与所述化合物1的总加料量的摩尔比为0.1-0.5:1;在一些优选的实施方式中,该摩尔比为0.2-0.4:1;在一些更优选的实施方式中,该摩尔比为0.25:1。
[0040]
在一些实施方式中,所述化合物1的总加料量与所述化合物2的总加料量的摩尔比为1-5:1;在一些优选的实施方式中,该摩尔比为1-1.2:1;在一些更优选的实施方式中,该摩尔比为1.05:1。
[0041]
在一些实施方式中,在所述分批加料的过程中,每批次加料的间隔时间为10-90分钟;在一些优选的实施方式中,所述间隔时间为30-75分钟;在一些更优选的实施方式中,所述间隔时间为60分钟。
[0042]
在一些实施方式中,所述反应压力为0.065-0.085mpa;在一些优选的实施方式中,所述反应压力为0.067-0.082mpa时,脱羧杂质和异构胺杂质的含量均小于0.5%。
[0043]
在一些实施方式中,所述与水不互溶的有机溶剂为芳香类溶剂,如氯苯、二甲苯或二氯苯。在非负压条件下,氯苯、二甲苯或二氯苯等高沸点溶剂的反应效果均较好;在负压条件下,氯苯和二氯苯的反应效果较好。
[0044]
在本发明中,溶剂的用量对反应的效果影响较小,在一些实施方式中,所述与水不互溶的有机溶剂与所述化合物2的总加料量的摩尔比为2:1以上。
[0045]
在一些实施方式中,所述加热的温度为110-130℃;在一些优选的实施方式中,所述加热的温度为118-125℃。
[0046]
在一些实施方式中,反应压力为常压,且化合物2的加料方式为分批加料时,将反应温度设置为130℃,所述化合物1的总加料量与所述化合物2的总加料量的摩尔比为1.2:1,可将脱羧杂质和异构胺杂质的含量控制在0.5%以下,反应的转化率和收率也较高。
[0047]
在一些实施方式中,反应压力为0.065-0.085mpa(如0.067mpa),且化合物2的加料方式未采用分批加料方式(如采用一次性加料方式),将反应温度设置为118℃,所述化合物1的总加料量与所述化合物2的总加料量的摩尔比为1.05:1时,可将脱羧杂质和异构胺杂质
的含量控制在0.5%以下,反应的转化率和收率也较高。
[0048]
在一些实施方式中,所述制备方法还可以包括后处理的步骤。
[0049]
在一些实施方式中,上述反应结束后,降温,加入盐酸溶液,分相,取有机相,即得化合物3的溶液,如化合物3的氯苯溶液。该化合物3的溶液可以直接参加后续制备唑啉草酯的反应。
[0050]
在一些实施方式中,当溶剂为氯苯时,上述反应结束后,降温,加入盐酸,过滤,所得固体即为化合物3。在一些实施方式中,所述降温的温度可以为20℃以下,所述盐酸的摩尔量与化合物2的总加料量的摩尔量相同。在本发明的实施例中,反应结束后监测反应液中化合物3的hplc相对含量,采用该后处理方法得到化合物3的绝对收率大致比相对含量降低2-3%。
[0051]
另外,上述唑啉草酯中间体的制备方法还可应用于唑啉草酯的合成,具体地,本发明还提供了一种唑啉草酯的制备方法,其包括以下步骤:
[0052][0053]
其中,化合物3由上述唑啉草酯中间体的制备方法得到。
[0054]
在本发明中,化合物1可以通过以下反应获得:
[0055][0056]
化合物4在氯苯为溶剂的条件下,通过利用一定浓度的氢氧化钾进行水解,反应结束后,分相,收集化合物1的氯苯溶液,化合物1的氯苯溶液可直接用于下一步反应,即化合物3的合成中。
[0057]
实施例
[0058]
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0059]
实施例1
[0060]
称取化合物2(hplc纯度98%,5.96g,0.0235mol)和化合物1的氯苯溶液(质量浓度5%,57.5g,0.0282mol)加入到100ml的三口瓶中,开始加热,将反应温度保持在130℃,反应5h后,停止反应,hplc检测相对含量,其中:化合物3为96%,脱羧杂质为0.8%,异构胺杂质为0.6%,化合物2<0.1%。
[0061]
实施例2-5
[0062]
按照实施例1的方式,改变反应温度,结果如下表1所示。
[0063]
表1
[0064][0065]
由表1可知:将反应原料一次性加入,在常压下反应,即使改变反应温度,脱羧杂质和异构胺杂质的含量仍然较高,并且原料的转化率显著降低。
[0066]
实施例6
[0067]
化合物1的氯苯溶液(质量浓度5%,28.5g,0.0282mol)加入到100ml的三口瓶中,保持反应体系处于常压的反应压力条件,开始加热,将反应温度保持在130℃后,利用固体分批加料器分四批加入化合物2(hplc纯度98%,5.96g,0.0235mol),每批化合物2加入量为1.49g。其分批加入的具体方式为:加入第一批化合物2,反应1h;再加入第二批化合物2,反应1h;再加入第三批化合物2,反应1h;最后加入第四批化合物2,反应1h后,停止反应。反应共计4h,hplc检测相对含量,其中:化合物3为98%,脱羧杂质为0.3%,异构胺杂质为0.5%,化合物2<0.1%。
[0068]
实施例7
[0069]
按照实施例6的方式进行,将反应温度设置为110℃。hplc检测相对含量,其中:化合物3为85%,脱羧杂质为0.3%,异构胺杂质为0.3%,化合物2剩余11%。
[0070]
在反应温度110℃下继续延长反应时间至15h,hplc检测相对含量,其中:化合物3为93%,脱羧杂质为0.6%,异构胺杂质为0.7%,化合物2剩余2%。
[0071]
实施例8
[0072]
化合物1的甲苯溶液(质量浓度5%,28.5g,0.0282mol)加入到100ml的三口瓶,开始加热,将反应温度保持在110℃后(甲苯回流),利用固体分批加料器分四批加入化合物2(hplc纯度98%,7.00g,0.0282mol),每批化合物2加入量为1.75g。其分批加入的具体方式为:加入第一批化合物2,反应1h;再加入第二批化合物2,反应1h;再加入第三批化合物2,反应1h;最后加入第四批化合物2,反应1h后hplc检测相对含量,其中:化合物3为73.6%,脱羧杂质为0.44%,异构胺杂质为0.2%,化合物2剩余25%。
[0073]
在回流温度下继续延长反应时间至12h,反应结束,hplc检测相对含量,其中:化合物3为96%,脱羧杂质为0.6%,异构胺杂质为0.8%,化合物2<0.1%。
[0074]
实施例9-11
[0075]
实施例9和10是按照实施例6的方式进行,设置不同的化合物1与化合物2的摩尔比。
[0076]
实施例11是按照实施例7的方式进行,设置反应温度为120℃。
[0077]
反应结果如表2所示。
[0078]
表2
[0079][0080]
由表2可知:在分批加料的方式下,脱羧杂质显著减低。化合物1与化合物2的摩尔比越高,反应结束后化合物3的含量越高,脱羧杂质和异构体杂质的含量稍有下降,但是化合物1与化合物2的摩尔比越高,就会导致化合物1的原子利用率不高,造成物料的浪费。
[0081]
实施例12
[0082]
化合物1的氯苯溶液(质量浓度5%,28.5g,0.0282mol)加入到100ml的三口瓶中,保持反应体系处于0.082mpa的反应压力条件,开始加热,将反应温度保持在125℃后,利用固体分批加料器分四批加入化合物2(hplc纯度98%,6.8g,0.0268mol),每批化合物2的加入量为1.7g。其分批加料的具体方式为:加入第一批化合物2,反应1h;再加入第二批化合物2,反应1h;再加入第三批化合物2,反应1h;最后加入第四批化合物2,反应1h后,停止反应。反应共计4h,hplc检测相对含量,其中:化合物3为98.5%,脱羧杂质为0.3%,异构胺杂质为0.2%,化合物2<0.1%。
[0083]
实施例13
[0084]
称取化合物2(hplc纯度98%,6.8g,0.0268mol)和化合物1的氯苯溶液(质量浓度5%,28.5g,0.0282mol)加入到100ml的三口瓶中,保持反应体系处于0.067mpa的反应压力条件,开始加热,将反应温度保持在118℃,反应4h后,停止反应,hplc检测相对含量,其中:化合物3为97.2%,脱羧杂质为0.5%,异构胺杂质为0.3%,化合物2<0.1%。
[0085]
实施例14-15
[0086]
按照实施例12的方式,设置不同的负压条件及反应温度,结果如表3所示。
[0087]
表3
[0088][0089]
由表3可知:控制反应压力在0.067mpa至0.082mpa之间,温度控制在120℃左右,反应的条件为最优条件,可以有效地将脱羧杂质和异构胺杂质同时控制在0.5%以下,同时化合物2的转化率也较高,化合物3的含量也在97%以上。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1